Proteomic profiles and ultrastructure of regenerating protoplast of Bryopsis plumosa (Chlorophyta)
Article information
Abstract
When a multinucleate cell of Bryopsis plumosa was collapsed by a physical wounding, the extruded protoplasm aggregated into numerous protoplasmic masses in sea water. A polysaccharide envelope which initially covered the protoplasmic mass was peeled off when a cell membrane developed on the surface of protoplast in 12 h after the wounding. Transmission electron microscopy showed that the protoplasmic mass began to form a continuous cell membrane at 6 h after the wounding. The newly generated cell membrane repeated collapse and rebuilding process several times until cell wall developed on the surface. Golgi bodies with numerous vesicles accumulated at the peripheral region of the rebuilding cell at 24 h after the wounding when the cell wall began to develop. Several layers of cell wall with distinctive electron density developed within 48–72 h after the wounding. Proteome profile changed dramatically at each stage of cell rebuilding process. Most proteins, which were up-regulated during the early stage of cell rebuilding disappeared or reduced significantly by 24–48 h. About 70–80% of protein spots detected at 48 h after the wounding were newly appeared ones. The expression pattern of 29 representative proteins was analyzed and the internal amino acid sequences were obtained using mass spectrometry. Our results showed that a massive shift of gene expression occurs during the cell-rebuilding process of B. plumosa.
INTRODUCTION
Some coenocytic green algae are able to rebuild a new cell from the remnants of destroyed cell in sea water (Tatewaki and Nagata 1970, Kobayashi and Kanaizuka 1985, Pak et al. 1991, Kim and Klochkova 2004, Bottalico et al. 2008). Several very distinctive sequential steps of this cell rebuilding process have been revealed from detailed cellular and molecular studies: agglutination of cell organelles into protoplasmic masses in sea water, formation of a primary polysaccharide envelope on their surface, replacement of this primary envelope by a lipid-based plasma membrane, and formation of a cell wall (Kim et al. 2001, 2002).
The generation of cell membrane on the surface of agglutinated protoplasmic mass is the most crucial event of the cell rebuilding process in Bryopsis plumosa (Hudson) Agardh. Previous study using Nile Red as fluorescent probe for intracellular lipids showed that formation of cell membrane occurred during the first 6 h from the moment of protoplasm extrusion into seawater (Kim et al. 2001). During this whole time period, the primary polysaccharide envelope that enclosed protoplasts acted like a cell membrane, showing semi-permeability and selective transport of materials. Calcofluor White staining showed that cell wall development occurred in 24–48 h after the wounding (Kim et al. 2001, 2002, Klochkova et al. 2003). Ultrastructure of the regenerating protoplast of B. plumosa has been reported (Pak et al. 1991), but detailed regeneration processes of cell membrane and cell wall are still unknown. As each step of protoplast formation is very distinct in terms of time intervals and requires a cascade of specific cellular events, one can expect that different groups of genes are regulated at each step.
Proteomics is one of the most powerful tools in modern biology to study the differential display of gene expression since proteins are often directly related to biological processes and cellular function (Pandey and Mann 2000, Tyers and Mann 2003). Two-dimensional polyacrylamide gel electrophoresis (2-DE), which can separate complex protein mixtures, was used to identify protein changes that occur normally during development and tissue differentiation, as a result of disease, drug treatment, environmental change, etc. (Celis et al. 1999a, 1999b, Spandidos and Rabbitts 2002). In phycology, this branch of “omics” has been widely applied, as evident from a large number of publications accumulating recently (e.g., Kim et al. 2008, Choi et al. 2015). Proteomic approach has been applied in Bryopsis studies by Yamagishi et al. (2004) who performed 2-DE analysis of the hybrid plants that developed from hybrid protoplasts of B. plumosa obtained by mixing protoplasm of its gametophyte and sporophyte. Most proteins synthesized by the hybrid plants were identical to those of normal sporophytes of B. plumosa (Yamagishi et al. 2004). As they used mature plants for the proteomic study, the changes of proteome profiles at early stage of cell rebuilding was not observed.
Ultrastructure of rebuilding cell was observed over a time course to determine exact timing of cell membrane regeneration and cell wall development in B. plumosa. To study proteome profiles during the cell rebuilding process, we constructed 2-DE protein maps before and after collapse of its giant cell. Proteins which showed significant expression changes at each stage were chosen and characterized using matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS).
MATERIALS AND METHODS
Plant material and laboratory culture
Vegetative plants of B. plumosa were collected from Kachon, southern coast of Korea and maintained as unialgal cultures in IMR medium (Kim et al. 2006). Plants were grown at 23°C with 12 : 12 h L : D cycle and 15 μmol m−2 s−1 light intensity. Protoplasts were prepared as described in Kim et al. (2001) and grown at the same culture conditions as above.
Sample preparation for protein extraction
To understand the changes of proteome profiles during cell rebuilding process five 2-DE maps were prepared using following samples: 1) control vegetative plants before wounding (0 h); 2) 3-h-old protoplasts: the time when membrane “patches” begin to incorporate into a primary polysaccharide envelope; 3) 12-h-old protoplasts: plasma membrane development is complete; 4) 24-h-old protoplasts: cell wall formation is in progress; and 5) 48-h-old protoplasts: cell wall is complete and cell begins to show polarity. Culture medium was removed from the algal thalli (2 g) with a tissue paper and they were frozen in liquid nitrogen and used for protein extraction.
The wet weight of algal thalli used for protoplast preparations was 36 g. Protoplasts were prepared as described in Kim et al. (2001) and grown in 100 × 10-mm glass Petri dishes containing IMR medium with 50 mg L−1 ampicillin to avoid bacterial contamination. Protoplasts were harvested at 3, 12, 24, and 48 h after formation using a fine glass pipette, transferred to 1-mL plastic tubes and centrifuged at 1,500 ×g for 10–15 sec. IMR medium was removed from the tubes and precipitated protoplasts were frozen in liquid nitrogen and stored at −75°C. We avoided mechanical disruption of the protoplasts during harvesting and centrifugation procedures as much as possible.
To prepare samples for 2-DE, materials were washed twice in ice-cold phosphate buffer saline (PBS) and lysed in sample buffer, composed of 7 M urea (Sigma, St. Louis, MO, USA), 2 M thiourea (Sigma), 4% (w/v) 3-(3-cholamidopropy) dimethymmonio-1-propanesulfonate (CHAPS, Sigma), 1% (w/v) dithiothreitol (DTT, Sigma), 2% (v/v) pharmalyte (Amersham Biosciences, Uppsala, Sweden), and 1 mM benzamidine (Sigma). Proteins were extracted for 1 h at room temperature with vortexing. After centrifugation at 15,000 ×g for 1 h at 15°C, insoluble material was discarded and soluble fraction was used for 2-DE. Protein loading was normalized by Bradford assay (Bradford et al. 1976).
Two dimensional electrophoresis
Immobilized pH gradient dry strips were equilibrated for 12–16 h with 7 M urea, 2 M thiourea, containing 2% CHAPS, 1% DTT, and 1% pharmalyte, and were then loaded with 200 μg of sample. Isoelectric focusing was performed at 20°C using a Multiphor II electrophoresis unit and EPS 3500 XL power supply (Amersham Biosciences) following manufacturer’s instruction. The voltage was linearly increased from 150 to 3,500 V during 3 h for sample entry followed by constant 3,500 V, with focusing complete after 96 kVh. Prior to the second dimension, strips were incubated for 10 min in equilibration buffer (50 mM Tris·Cl buffer of pH 6.8, 6 M urea, 2% sodium dodecyl sulfate [SDS], and 30% glycerol; Sigma), first time with 1% DTT and second time with 2.5% iodoacetamide (Sigma). Equilibrated strips were inserted onto sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels (20–24 cm, 10–16%). SDS-PAGE was performed using Hoefer DALT 2-DE system (Amersham Biosciences) following manufacturer’s instruction. 2-DE gels were run at 20°C for 1.7 kVh. Thereafter, the gels were silver-stained as described by Oakley et al. (1980), except that fixing and sensitization step with glutaraldehyde were omitted. Minimum three copies using total proteins extracted from different material were analyzed for the proteome profiles of each stage.
Image analysis
Quantitative analysis of digital images was carried out using the PDQuest software (version 7.0; BioRad, Hercules, CA, USA) according to the manufacturer’s protocols. Quantity of each spot was normalized by total valid spot intensity. Protein spots were selected for the significant expression variation deviated over two fold in its expression level, compared with control or normal sample.
Enzymatic digestion of protein in-gel
Protein spots were enzymatically digested in-gel in a manner similar to that described by Shevchenko et al. (1996), using modified porcine trypsin (Promega, Madison, WI, USA). Gel pieces were washed with 50% acetonitrile (Sigma) to remove SDS, salt and stain, dried to remove the solvent and then rehydrated with trypsin (8–10 ng μL−1) and incubated for 8–10 h at 37°C. The proteolytic reaction was terminated by addition of 5 μL of 0.5% trifluoroacetic acid (Sigma). Tryptic peptides were recovered by combining the aqueous phase from several extracted gel pieces with 50% aqueous acetonitrile. After concentration, the peptide mixture was desalted using C18ZipTips (Millipore Co., Bedford, MA, USA), and peptides were eluted in 1–5 μL of acetonitrile. An aliquot of this solution was mixed with an equal volume of a saturated solution of α-cyano-4-hydroxycinnamic acid (Sigma) in 50% aqueous acetonitrile, and 1 μL of mixture was spotted onto a target plate.
Mass spectrometry determination and similarity searches
Protein analysis was performed using an Ettan MALDI-TOF MS (Amersham Biosciences). Peptides were evaporated with a N2 laser at 337 nm using a delayed extraction approach. They were accelerated with 20 kV injection pulse for time-of-flight analysis. Each spectrum was the cumulative average of 300 laser shots. Spectra were calibrated with trypsin auto-digestion ion peak m/z (842.510, 2,211.1046) as internal standards. The search program ProFound, developed by the Rockefeller University (http://prowl.rockefeller.edu/prowl-cgi/profound.exe) was used for protein identification by peptide mass fingerprinting (Zhang and Chait 2000). These procedures were performed in Korea Basic Science Institute (KBSI, Daejeon, Korea). Additional search was performed using publically available BLASTp (protein-protein BLAST) option in the National Center for Biotechnology Information (NCBI) database (NCBI 2016).
Microscopy
For time-lapse video microscopy, protoplasts were placed on a glass slide and a coverslip was lowered and sealed with VALAP (1 : 1 : 1; vaseline/lanolin/paraffin) melted on a hot plate at 70°C. The slide preparations were examined and recorded using a digital imaging time-lapse recorder (AllBT, Daejeon, Korea) mounted on the microscope.
For transmission electron microscopy (TEM), protoplasts were prepared on 2% agar. The protoplasts partially embedded on agar gels were fixed in specially developed fixative solution (1% para-formaldehyde, 2.5% glutaraldehyde, 2.5% dimethyl sulfoxide, 10% sucrose, and 0.05% CaCl2 in 0.5 M cacodylate buffer) for 2 h. Samples were then rinsed with the same buffer and post-fixed with 1% osmium tetroxide at 4°C for 2 h. Thereafter, they were rinsed out with PBS buffer and were dehydrated in a graded ethanol series with 10% increments (each step was 10 min), embedded in Spurr’s epoxy resin and polymerized overnight at 72°C oven (Polysciences Inc., Warrington, PA, USA). Resin-embedded samples were sectioned with a diamond knife, and thin sections stained with uranyl acetate for 30 min and 2% (w/v) lead citrate for 10 min were viewed and photographed on a JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan).
RESULTS
When the protoplasm was extruded from the cut-open B. plumosa thalli into seawater, the cell organelles agglutinated into small protoplasmic masses at first. During the next 10–20 min each protoplasmic mass contracted, became spherical and enclosed by a primary polysaccharide envelope, forming a compact protoplast-like structure (Fig. 1A–D). This primary protoplast repeatedly ruptured and regenerated the structure during the first 3 h with active rotation of the cell organelles. Observations with time-lapse video microscopy showed that not only the primary envelope but also the newly generated plasma membrane collapsed at least 2–3 (6) times during the first 12 h after protoplast formation (Fig. 1E–L). Each time it reformed the membrane again; however, activity to reform slowed down each time, and some protoplasts could not reform anymore and degenerated.
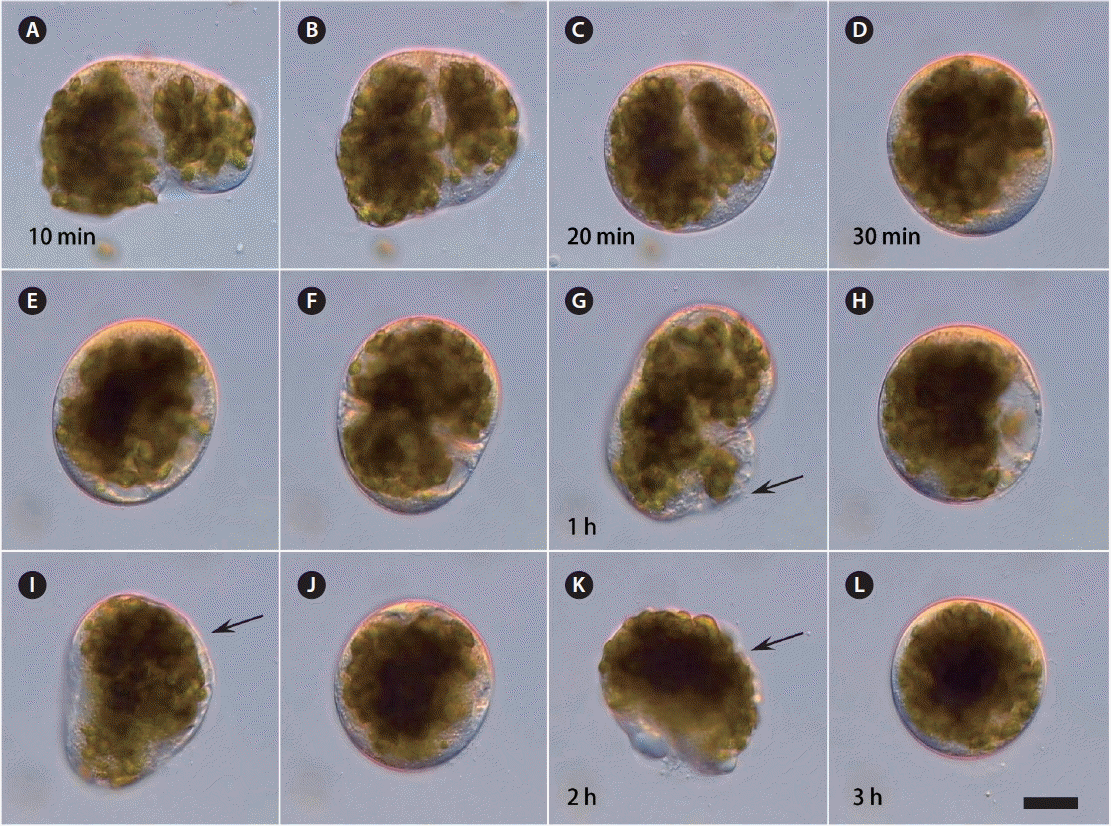
Time-lapse video microscopic observations on Bryopsis plumosa protoplast during the first 3 h after formation from the extruded protoplasm in seawater. (A–L) Primary polysaccharide envelope can break and re-form 2–3 times during the first 2 h (arrows). Scale bar represents: 10 μm.
TEM showed that some discontinuous “patches” of a membrane began to appear on the surface of protoplasmic mass at 3 h after its formation (Fig. 2A), and complete plasma membrane was formed by 6–12 h after the wounding (Fig. 2B & C). Previously, Calcofluor White staining showed that accumulation of cell wall material started at 6–8 h after protoplast formation (Kim et al. 2001), but distinctive ultrastructure of cell wall was first observed at 24 h after the wounding. An accumulation of Golgi bodies and numerous vesicles was observed along the peripheral region of the protoplast at 20–24 h after the wounding. The deposited cell-wall material became more distinguishable on ultrathin sections, and a layer of materials with numerous vesicles was clearly visible on the surface of the developing protoplast (Fig. 2D). After 48 h, multilayered thick cell wall (2–2.5 μm) developed. Each layer of the cell wall showed different electron density suggesting that the developing cell deposited different types of cell-wall materials during the rebuilding process (Fig. 2E & F).
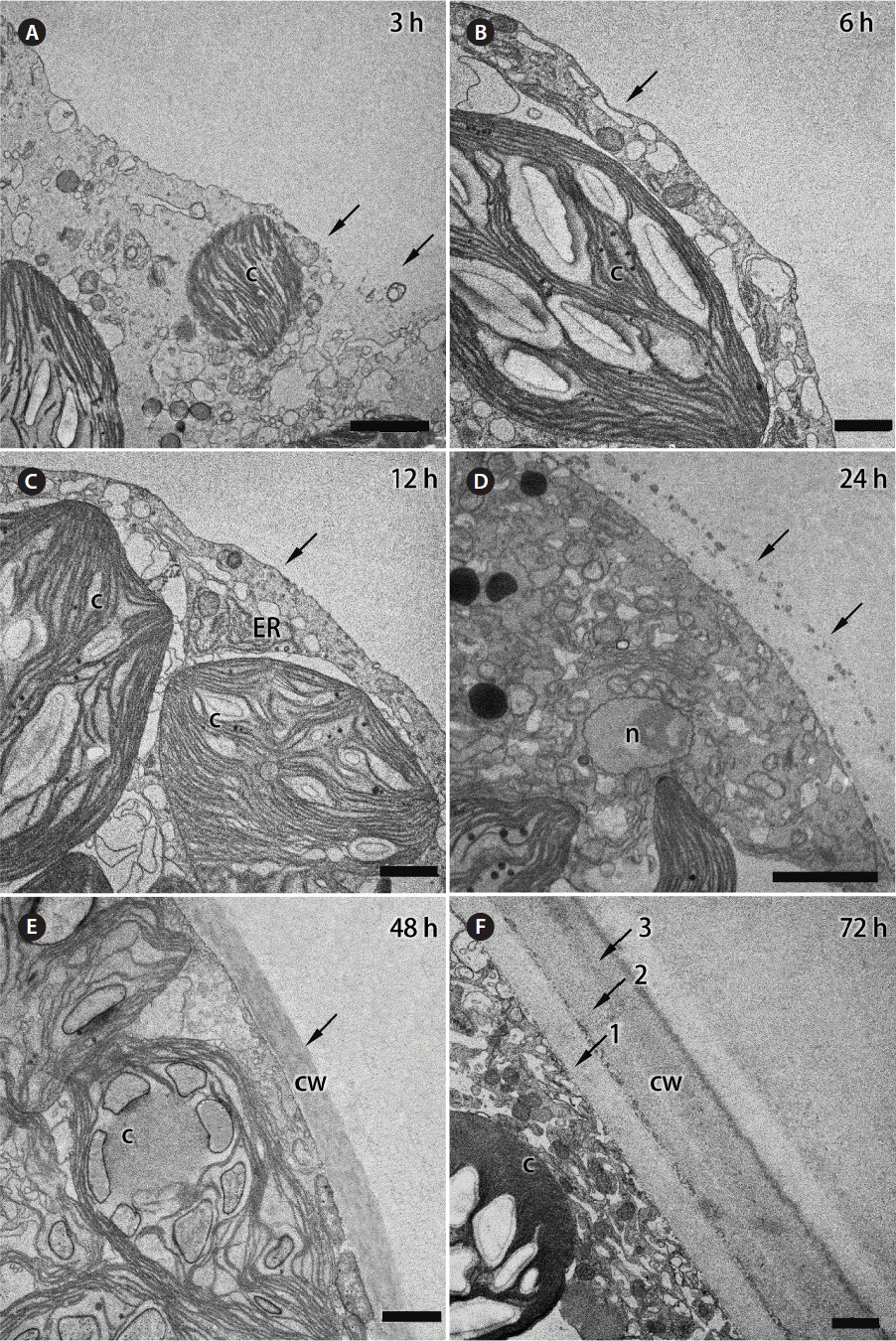
Transmission electron micrographs of Bryopsis plumosa protoplasts showing changes of their outer surface structure per different times after formation. (A) During the first 1 h after formation, protoplast is not covered with a lipid-based cell membrane and it is enclosed with an amorphous polysaccharide envelope. By 3 h after protoplast formation, some discontinuous membranous “patches” began to appear on its surface, which are interrupted with areas without membrane (arrows). (B & C) The plasma membrane is fully formed in the period between the 3rd and 12 h of the protoplast’s life (arrows). (D) A thin cell wall is visible on the surface of protoplast at 24 h after its formation (arrows). (E) One-layered cell wall at 48 h after protoplast formation in seawater (arrow). (F) Thick three-layered cell wall at 72 h after protoplast formation. Three distinct layers are shown with arrows and numbers. c, chloroplast; cw, cell wall; ER, endoplasmic reticulum; n, nucleus. Scale bars represent: A & D, 2 μm; B, C, E & F, 1 μm.
Protein profile in vegetative plants, protoplasts, and newly formed cells
Proteome profiles of B. plumosa changed dramatically in short time intervals during the cell rebuilding process (Fig. 3). Most proteins that appeared in control disappeared during the process and many new proteins specifically appeared at each stage of protoplast formation (Fig. 4). The silver-stained gels showed 1,350–1,420 protein spots in control (0 h), but only 259 proteins were detected throughout the cell rebuilding process (3–48 h). Quantitative analysis showed that 59.8% of the proteins that appeared in control were significantly (<3 fold change) down-regulated and 10.8% of the proteins were up-regulated in 24-h-old protoplasts. About 70–80% of proteins detected at 48 h were not detected at all in the initial stage (0–12 h). As a result, the proteome profile at 48 h after the wounding was almost completely different from those of control and early stage of protoplast formation (Fig. 3). The number of detected proteins increased over time course; total number of detected proteins at 24 and 48 h was 1,860 and 1,915 spots, respectively; 25–36% increase compared to those in control (1,420 spots) and in 3 h (1,234 spots). These results (dramatic change in proteome profile and increase of detected protein spots in late stage of protoplast formation) were repeated three times using different materials. It is noteworthy that the protein spots strongly expressed in early stage disappeared over time course and the proteome profile of 48-h-old protoplasts was filled with numerous weak protein spots (Fig. 3).


Enlarged images, showing comparison of intensities in the expression of some representative differentially expressed proteins detected on the proteomic maps of the vegetative plants of Bryopsis plumosa (0 h) and protoplasts at 3, 12, 24, and 48 h after formation. (A) Protein spot 2211 identified as chaperone protein pmfD precursor. (B) Protein spot 6617 (unidentified). Mr and pI value of that protein spot were 67.6 and pH 5.5, respectively. (C) Protein spots 8110 and 8209 identified as alanine racemase and glycoprotein UL9, respectively.
Internal amino acid sequence was obtained from 49 chosen proteins which were specifically up-regulated at each cell-rebuilding stage and 29 proteins were annotated (Table 1). Only 21 proteins were annotated with >80% similarity, and 6 proteins were annotated the same with two approaches, ProFound program and BLASTp. Most identified proteins belonged to catalytic enzymes involved in ordinary cell metabolism (Table 1). In general, the number of analyzed proteins was too small to understand any proteome profiles in detail.

Expression level and molecular characteristics of some differentially displayed proteins extracted from 2-DE gels of the vegetative plants (0 h and control), protoplasts (3, 12, and 24 h), and newly formed cells (48 h) of Bryopsis plumosa
DISCUSSION
Cell rebuilding process of Bryopsis plumosa is composed of several distinctive events, and development of plasma membrane and cell wall are the most crucial steps to complete the process (Kim et al. 2001). The exact time of each developmental step was determined for the first time using ultrastructural analysis. Observations with time-lapse video microscopy showed that the rebuilding cell may undergo the collapse of cell membrane at least 2–3 (6) times during the first 12 h. A comparative proteomic experiment was designed based on these times, and the results revealed a striking shift of proteome profile before and after the development of cell membrane. About 60% of proteins detected in control vegetative cell disappeared or significantly reduced at late stage of cell rebuilding, and about 70–80% (over 1,000 spots) of proteins detected at 48 h were newly appeared. Such a massive proteomic shift is very rare in any organisms. The male and female gametophytes of red algae, Bostrychia radicans and B. moritziana, show only 5–48 differential proteins out of 699–921 total protein spots (Kim et al. 2008). The proteome shift observed during cell-rebuilding of B. plumosa is similar to that reported in germinating seeds of higher plants which is driven by an epigenetic modification (Gallardo et al. 2001).
Time-lapse video microscopy showed that there was an active relocation of cell organelles inside protoplasmic mass surrounded with primary envelop. This active movement of protoplasmic mass often caused the collapse of developing protoplasts at early stage of cell rebuilding process. There are very few ultrastructural studies on cell rebuilding process of B. plumosa because of difficulties in fixation of this fragile structure (Pak et al. 1991). We could stably fix the structure of regenerating protoplast using a fixative containing 10% sucrose and agar-embedding method, which enabled us to determine the exact time of cell membrane and cell wall development. The protoplasmic mass was surrounded with a continuous cell membrane at 6 h after the wounding, but the collapse and rebuilding of developing protoplast was observed until 12 h after the wounding, suggesting that there are mechanisms holding the cell organelles together until whole cell-rebuilding process is complete with the development of cell wall.
A lectin involved in the aggregation of the extruded cell organelles in seawater and initial formation of the protoplasmic mass was isolated from B. plumosa and named as bryohealin (Kim et al. 2006, Yoon et al. 2008). This lectin is universal not only for other populations of B. plumosa (NCBI accession No. BAI43481), but also for other species of Bryopsis, as it was found in B. hypnoides Lamouroux (Xu et al. 2012) and B. maxima Okamura ex Segawa (NCBI accession No. BAI94586). Bryohealin contains an antibiotic domain and its function may be protection of the newly generated protoplasts from bacterial contamination (Klochkova et al. 2005, Yoon et al. 2008). Several additional lectins involved in the aggregation of expelled cell organelles were isolated from B. plumosa and confirmed that a lectin-carbohydrate complementary system mediates agglutination of extruded cell organelles (Han et al. 2010, 2011, 2012, Jung et al. 2010). Niu et al. (2009) suggested that the aggregation of cell organelles might be proton-gradient dependent, because it was inhibited by nigericin. Xu et al. (2012) reported that a 43 kDa lectin appeared at 3 h after protoplasm extrusion into seawater and then disappeared at 6 h from western blotting analysis. These data are confusing, because the amino acid sequence of their “novel” lectin from B. hypnoides is 100% identical to bryohealin from B. plumosa, clearly implying these are the same proteins. Bryohealin has the molecular weight of 54 kDa and it usually shows little migration change on 2-DE gel (Kim et al. 2006, Yoon et al. 2008). It is more likely that the regenerating protoplast is using lectins which were produced before the cell rupture to hold cell organelles together until cell wall development.
The ultrastructure of cell wall on rebuilt protoplast of B. plumosa was observed for the first time. Golgi bodies with numerous vesicles were observed at the peripheral region of the rebuilding cell at 24 h after the wounding. Cell wall showed distinctive layers with different electron density at 48–72 h after the wounding. Previous studies showed that the newly built cell showed polarity at 48 h after the wounding (Kim et al. 2001, Kim and Klochkova 2004). It may be a coincidence, but further investigation using this cell-rebuilding model system may reveal the relationship between the development of cell wall and the occurrence of polarity in developing cell.
Our results showed the “pros and cons” of proteomic methods. The ‘pros’ of proteomic methods are simple and fast profiling of gene expression on a 2-DE gel. The general change of proteome profile over time course was clearly demonstrated on each 2-DE gel and the pattern was easy to understand at a glance. However, detailed analysis on differentially displayed proteins was disappointing. The ‘cons’ of proteomic methods are the difficulty in getting amino acid sequence from each protein spots (Kim et al. 2008). Although proteomics is a powerful tool to study the differentially expressed proteins involved in various biological processes, it offers very limited access to the actual characteristics of weakly expressed proteins (Pandey and Mann 2000, Tyers and Mann 2003). Less than 5% of protein spots observed in silver-enhanced gel are useful for the analysis using mass spectrometer. We did not expect that such a large scale of proteome changes would occur during the cell rebuilding process. Less than 3% of total protein spots had enough amounts to be isolated from the gels and were good for identification using mass spectrometry. Therefore, 29 proteins that we could analyze further with mass spectrometry were not enough to represent the specific gene regulation at each stage of cell rebuilding process. It seems not worthy to discuss possible role of the identified proteins in protoplast formation except for two chaperones that were up-regulated at the early stage of protoplast formation. The common feature of lipase chaperone, also called as lipase foldase or lipase activator protein, is to assist in the protein folding. They are also involved in the translocation of proteins across the membranes (Smith et al. 1998, Feldman and Frydman 2000), rearrangement of disulfide bonds in reduced proteins, and vesicular trafficking and signal transduction (Aitken 1996). Many chaperones are heat shock proteins and their expression begins in response to various stresses (Ellis and van der Vies 1991). Considering that new cell membrane develops at the early stage (3–12-h-old protoplasts), the up-regulation of many chaperons at this time is expected. Our results showed that cell-rebuilding process of B. plumosa is accompanying a large scale of genetic shift at each developmental stage. Transcriptomic analysis based on deep sequencing will be more useful in understanding molecular features of these complicated cell-rebuilding steps.
ACKNOWLEDGEMENTS
This work was supported by a National Research Foundation of Korea Grant (NRF-2015M1A5A1041804) funded to GHK. This research was also supported by Golden Seed Project, Ministry of Agriculture, Food and Rural Affairs, and a grant from Marine Biotechnology Program (PJT200620, genome analysis of marine organisms and development of functional applications) funded by Ministry of Oceans and Fisheries, Korea.