INTRODUCTION
Cyanidioschyzon merolae is a eukaryotic, extremophilic, red alga which thrives at low pH (approximately 0.2 to 4) environment and moderately high temperature (40–56°C). In recent years, this organism has gained much attention thus becoming a new model eukaryote for cell and organelle biology research (Kuroiwa et al. 2017). The newly developed methods of genetic transformation of the nuclear (Zienkiewicz et al. 2018) and the chloroplast genome (Zienkiewicz et al. 2017b) might enable a broad range of possible research in physiology and biotechnology. The prospect of expression of recombinant proteins in the chloroplast has attracted considerable attention as chloroplast may accumulate considerable quantities of protein (Maliga 2004, Bock 2007, Daniell et al. 2009). The accumulation of protein in chloroplasts may reach as much as 70% of the total soluble protein in a leaf of the cell (Daniell et al. 2009, Zienkiewicz et al. 2017b). Introduction of exogenous genes expression into the chloroplast genome requires a host promoter, either constitutive or inducible. Until recently there were no reliably identified promoter sequences useful in chloroplast transformation.
Here, we have investigated the practical applicability of three selected chloroplast promoters for stable and highly efficient overexpression of recombinant proteins in C. merolae chloroplasts. We have selected three gene promoters, previously (Kanesaki et al. 2012) identified as dependent on the light irradiance (PpsbD–promoter of the photosystem II D2 protein, gene psbD [CMV081C]), dependent on the cell cycle (PrbcL–promoter of the Ribulose-1,5-bisphosphate carboxylase / oxygenase [RuBisCO] large chain, gene rbcL [CMV013C]) as well as mostly constitutive promoter (PdnaK of a Hsp70-type chaperone, gene dnaK [CMV163C]).
Chloramphenicol acetyltransferase (CAT) is a well-known (Shaw and Unowsky 1968) antibiotic resistance enzyme. It was first described as a resistance factor in Enterobacteriaceaea and its enzymatic activity is to facilitate catalysis deactivation of chloramphenicol by formation of 3-acetoxy chloramphenicol, a derivative incapable of binding with the 50S bacterial or chloroplast ribosome unit. Chloramphenicol blocks protein chain elongation by inhibiting the peptidyl transferase activity of the bacterial ribosome in the 23S rRNA of the 50S subunit. This antibiotic does not bind to eukaryotic ribosomes localized in cytosol nor in mitochondria due to lack of the 50S unit. However, chloroplast ribosomes do possess the 50S units considered to be similar enough, with those found in bacteria, to be potentially sensitive to chloramphenicol (Zienkiewicz et al. 2017b).
MATERIALS AND METHODS
Cell cultures
Cyanidioschyzon merolae, 10D (NIES-1332, unialgal, clonal, and non-axenic) strain was obtained from Microbial Culture Collection (http://mcc.nies.go.jp; Tsukuba, Japan) and was used throughout this study. Cells were grown in MA2 liquid medium (Minoda et al. 2004) in a glass vessel shaken at 100 rpm under continuous white light illumination (100 μmoles photon m−2 s−1) at 37°C or on Petri dishes filled with MA2 medium, solidified by addition of 0.4% gellan gum (Phytagel; Sigma-Aldrich, Munich, Germany) (Minoda et al. 2004) or 0.75% agar (Basica LE, Prona, EU).
Escherichia coli, strain DH5α (genotype: F- Φ80lacZΔM 15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK-, mK+) phoA supE44 λ-thi-1 gyrA96 relA1) were used for construction of transformation vectors (Hanahan 1983). Bacterial cells were cultured in liquid Luria-Bertani (LB) medium (1% Bacto tryptone, 0.5% yeast extract, 1% NaCl at 37°C) or on Petri dishes with LB medium solidified by addition 1% agar. For selecting of transformed cells, the medium was supplemented with Kanamycin (30 μg mL−1).
Escherichia coli competent cells
Preparation of E. coli competent cells and transformation was carried out as described earlier (Sambrock et al. 1989). Electrocompetent cells were prepared according to manufacturer instructions of GenePulser apparatus (Bio-Rad, Hercules, CA, USA).
Construction of plasmids
The detailed construction of the pCCATCH plasmid (12,455 bp, GeneBank accession No. KX056487) containing catCH gene, driven by dnaK promoter region (1,085 bp in size, coordinates 106,558–107,643 bp) has been previously described by Zienkiewicz et al. (2017b). The pCCATCHD (12,153 bp) and pCCATCHL (11,916 bp) are identical to pCCATCH with catCH gene driven by promoter regions of psbD gene (780 bp in size, coordinates 51,736–52,515 bp) or rbcL gene (543 bp in size, coordinates 6,960–7,503 bp), respectively. Similarly, as in the case of dnaK promoter, psbD and rbcL promoter regions were introduced into transformation plasmid molecules by pairs of primers with SalI and KpnI restriction enzymes sites. Coordinates refer to chloroplast molecule accession No. AB002583.1. The name of pCCATCH plasmid has been converted into pCCTACHK to indicate the use of the dnaK gene promoter region for catCH gene expresion.
Plasmid pCCATGN (13,770 bp) was used as a control in indirect immunofluorescence and fluorescence microscopy. Its construction was described before (Zienkiewicz et al. 2017a).
All primers used in this study were listed in Supplementary Table S1.
Enzymatic manipulations of DNA
All enzymatic manipulations of DNA, such as restriction digestion, blunting of cohesive termini using T4 polymerase, the Klenow fragment or ligation reaction were performed according to protocols supplied by the manufacturers.
Polymerase chain reaction
Polymerase chain reactions (PCRs) were performed according to manufactures protocols, supplied with the Phire Plant Direct PCR kit containing, Plant Phire Hot Start II DNA Polymerase or DreamTaq DNA polymerase (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Plasmid DNA purification and transformation of Cyanidioschyzon merolae
Plasmid DNA purification was performed with the Extractme Plasmid DNA Kit, Gdańsk, Poland. Polyethylene glycol (PEG)-mediated transfections were performed as described before (Ohnuma et al. 2008) with the following modifications. A freshly started, overnight grown 100 mL of C. merolae culture at OD750 0.4 was spun down at 2,000 ×g for 5 min in 40°C in a table-top centrifuge (5810 R; Eppendorf, Hamburg, Germany) and resuspended in 100 mL of MA-I buffer (20 mM [NH4]2SO4, 2 mM MgSO4, 1× trace elements at 40°C). Then spun down again and resuspended in 500 μL of MA-I. The transformation mixture was prepared as follows: 20 μg of the plasmid was diluted in 400 μL of the MA-I buffer and mixed with 100 μL of cell suspension, subsequently, 500 μL of 60% PEG (w/v) was added to final PEG concentration of 30% (w/v). The transformation mixture was incubated for 5 min at room temperature, then transferred to 50 mL of warm MA2 medium and incubated overnight in normal growth conditions. The following day the culture was spun down in 40°C at 2,000 ×g for 5 min and resuspended in 50 mL of fresh MA2 medium to wash off the remaining PEG. The culture was led for 3 days in normal growth conditions before the selectable conditions were introduced. On the fourth day, chloramphenicol was added to the final concentration of 150 μg mL−1. The selectable growth of transformed C. merolae culture was conducted for a further three months. Every seven days C. merolae cells were spun down (2,000 ×g for 5 min at 40°C) and resuspended in fresh MA2 medium supplemented with chloramphenicol (150 μg mL−1). After 21 days, the concentration of chloramphenicol was increased up to 200 μg mL−1. Further, the C. merolae cells were plated with a sterile paint brush on Petri dishes with solid MA2 (0.4% gellan gum) supplemented with chloramphenicol at the final concentration of 200 μg mL−1 and sealed with a triple layer of parafilm. Plates were incubated for at least four weeks until single colonies appeared. Single cell colonies (up to 10 colonies) were transferred to 20 mL flasks of MA2 medium with 200 μg mL−1 of chloramphenicol. The culture was led for several months with medium exchange every two weeks. In parallel, the same cultures were led in step-wise (extra 50 μg mL−1 for every step) increasing concentration of chloramphenicol.
Cyanidioschyzon merolae growth rate
All mutant strains and the wild-type (WT) were grown in a multicultuvator (PSI, Drasov, Czech Republic) and the grow-curves were recorded by the acquisition of the optic density at wavelengths of 680 and 720 nm at 30-min intervals. Cells were illuminated with an intensity of 100 μmoles photon m−2 s−1 of external fluorescent light for 168 h (7 days) in a continuous flow of sterile air 0.5 L min−1 and 37°C. Each curve was approximated with a sigmoidal function and the bending point was calculated as the time by which culture has reached half of the maximal optic density.
SDS-PAGE and immunoblotting analysis
Mutants and control cells were harvested by spinning down for 5 min at 2,000 ×g and resuspended in the resuspension buffer (20 mM Hepes-NaOH pH 7.6, 5 mM EDTA, and 330 mM sucrose). Cells were counted in Neubauer chamber at least 3 times. Cells samples were solubilized in denaturing buffer (0.25 M Tris-HCl [pH 6.8], 0,4% (w/v) sodium dodecyl sulfate, 10 M urea, 2% (v/v) 2-mercaptoethanol and 20% (v/v) glycerol) and mixed together in 1 : 1 (v/v) ratio. Proteins were separated on 15% gels by Laemmli-type sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) method (Laemmli 1970). Gel wells were loaded with 10 μL of cell lysate (obtained from 0.5 × 108 cells) and run under constant amperage (24 mA). Following electrophoresis, polypeptides were electro-transferred on PVDF-membrane (Towbin et al. 1979) and probed with rabbit anti-CAT (specific to CAT) antibodies (Sigma-Aldrich) or anti-D1 (Agrisera, Vännäs, Sweden). Bands specifically binding the probe were visualized by enhanced chemiluminescence method (SuperSignal West Pico; Thermo Scientific Inc.) according to standard procedures using ChemiDoc System (Bio-Rad).
Southern blot analysis
The transfer of the PstI digested DNA from the agarose gel to a nitrocellulose membrane was performed by a conventional alkaline method using 20× SSC buffer (3 M NaCl, 0.3 M sodium citrate, pH 7.0). DIG-labeled DNA fragments were prepared by PCR using DIG Probe Synthesis Kit (Roche, Basel, Switzerland) with specific primers (catgnF and catgnR hybridizing with the catgn sequence) and used as hybridization probes. The DIG was detected with alkaline phosphatase-conjugated anti-DIG antibody and CDP-Star (Roche). The signals were visualized with the luminescent image analyzer ChemiDoc XRS+ System (Bio-Rad).
Indirect immunofluorescence
C. merolae cells were grown and synchronized by 8 : 16 h light : dark regime. Cell fixation and permeabilization were performed as described before (Zienkiewicz et al. 2017a). Primary and secondary antibodies were used at the following concentrations: 1 : 100 for rabbit anti-CAT antiserum, 1 : 100 for Alexa-488 goat anti-rabbit antibody (Thermo Fisher Scientific Inc.).
Fluorescence microscopy
Cells were observed using microscope Olympus BX43F (Tokyo, Japan) with a set of filters: “DAPI” 49025–ET–DAPI/FluoroGold Longpass (Chroma, Bellows Falls, VT, USA) (excitation filter [EX] 300–400 nm, diachronic mirror [DM] > 400 nm, absorption filter [BA] > 425 nm) for blue fluorescence of DNA with Hoechst 33258 dye and red auto-fluorescence of chlorophyll at the same time. “Green/alexa anti-CAT” 49002–ET–EGFP (FITC/Cy2) (Chroma) (EX 450–490 nm, DM > 495 nm, BA 500–550 nm) for green fluorescence of Alexa 488 goat anti-rabbit antibody, “RED/chlorophyll” 39106–AT–Qdot 625 (Chroma) (EX 400–450 nm, DM > 575 nm, BA 590 nm) for red auto-fluorescence of chlorophyll, FITC / TRITC (EX 475–490 nm, DM > 485 nm, BA 610–640 nm) for red auto-fluorescence of chlorophyll.
RESULTS
Construction of transformation vectors and transformation of Cyanidioschyzon merolae
Selected promoter sequences were cloned into a transformation vector according to an established scheme (Fig. 1A). Correct introduction of promoter sequence was confirmed with basic genetic methods (Fig. 1B & C). Correct clones were confirmed by PCR and Southern blot analysis (Fig. 1D–F). A set of primers, used for construction of transformation vectors was provided in Supplementary Table S1.
The expression level of the exogenous protein
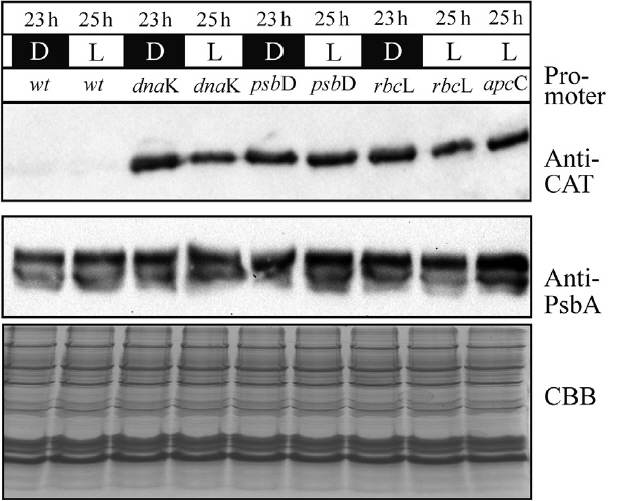
To assess the strength of all chloroplast promoters we have visualized the concentration of the overexpressed protein CAT (CAT, catCH gene) by immunoblotting (Fig. 2). All cell lines were synchronized by 2-day growth-period 6 L : 18 D (Kanesaki et al. 2012). After 15 h of darkness and after 1 h of light, cells were harvested, lyzed and analyzed by SDS-PAGE electrophoresis followed by western blotting. It was observed that the level of the CAT protein was present almost identical in all samples, independent of cell cycle or light irradiation.
Growth dynamics
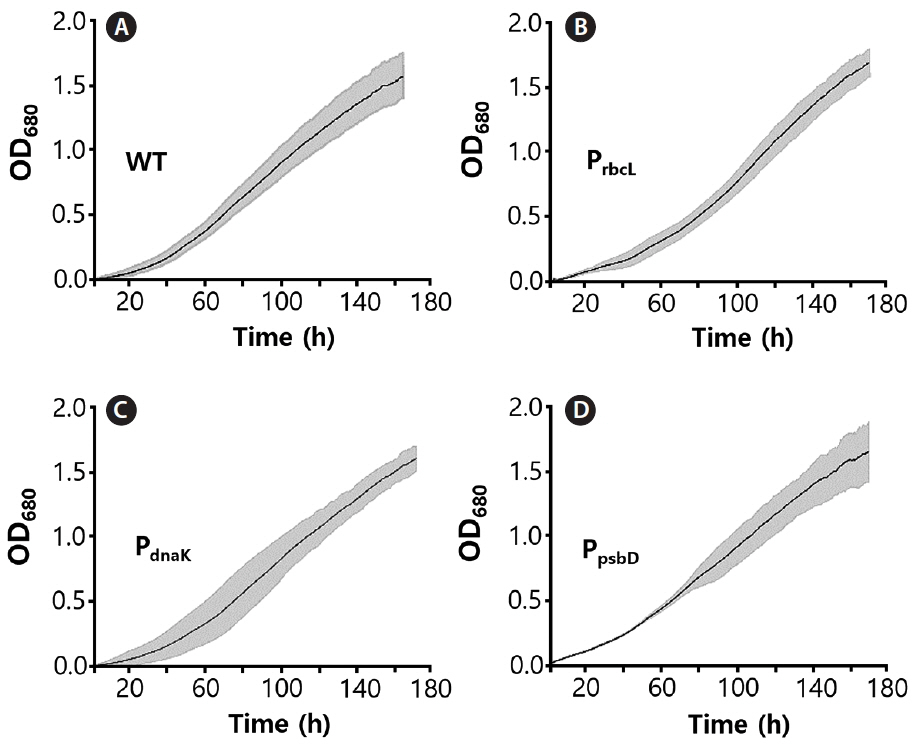
C. merolae cell growth curves were recorded continuously for seven days at irradiation of 100 μmoles photons m−2 s−1 in three repeats. It was observed that all mutants, as well as the WT, exhibited similar growth rate, suggesting that the resistance protein (CAT), expressed under all three promoters, provided complete protection from chloramphenicol (Fig. 3). All curves were approximated with a sigmoidal function and the bending point was calculated, as the time needed to achieve the maximal growth rate. It was observed that the WT cells, as well as all recombinant strains, grew at a nearly identical rate, achieving the bending point after 130 h (5.5 days) for the WT and up to 165 h (6.8 days) for the slowest (PsbD) strain (Fig. 4). The observed difference growth characteristic might be caused by metabolic exhaustion of mutant cells but it is likely to be insignificant. All mutants were grown 200 μg mL−1 chloramphenicol.
Subcellular location of the overexpressed proteins
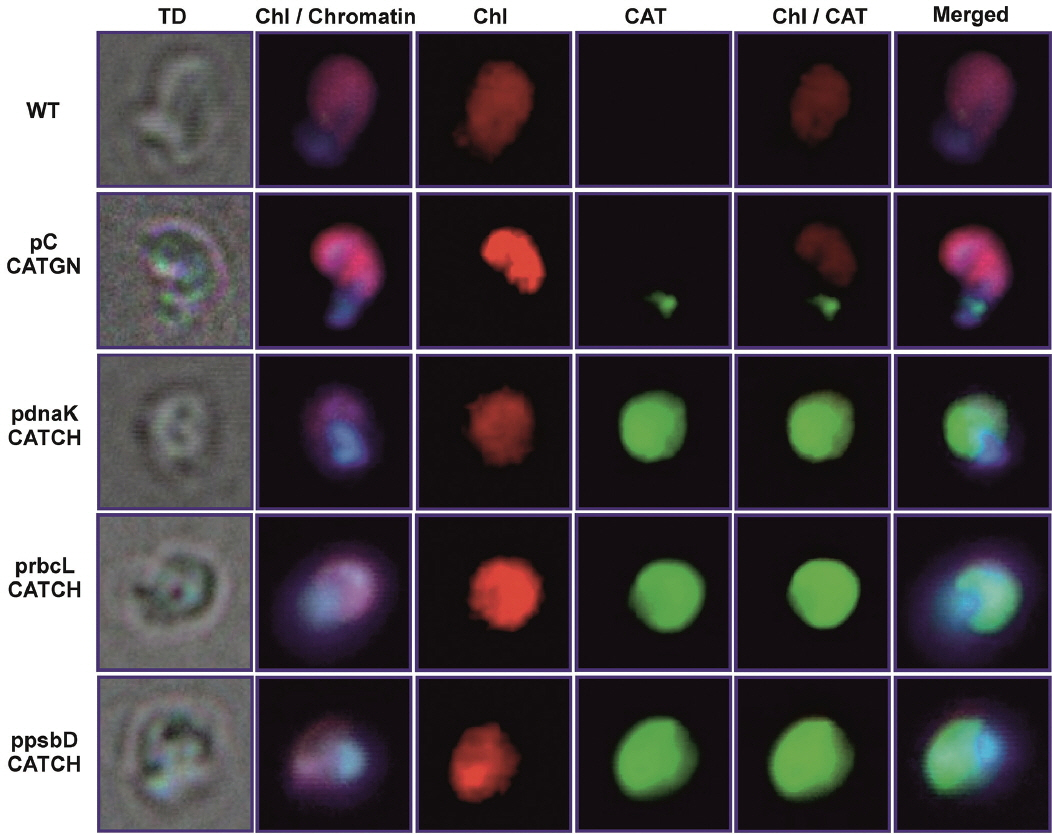
To confirm chloroplast location of the expressed CAT protein, mutant cell-samples were labeled with anti-CAT antibody and further with the fluorescent secondary antibody. It was observed that green fluorescence of labeled CAT protein always overlapped red fluorescence of chlorophyll. Additionally, the blue fluorescence of chromatin was never overlapped with chlorophyll nor with CAT location (Fig. 5).
DISCUSSION
We have successfully transformed C. merolae chloroplast genome, according to the method described before (Zienkiewicz et al. 2017b), thereby confirming its applicability. All the selected mutant stains exhibited nearly identical growth rate (Figs 3 & 4), confirming sufficient, if not complete, protection of the chloroplast biosynthetic machinery by the CAT protein from the toxic effects of chloramphenicol (Zienkiewicz et al. 2017a).
All three proposed promoter sequences are capable of delivering a highly stable and efficient chloroplast protein synthesis. The chloroplast location of CAT expression was confirmed by indirect fluorescence microscopy (Fig. 5). Despite the fact that the PpsbD promoter was reported to be highly dependent to light irradiation (Kanesaki et al. 2012), allowing for a highly variable transcription of the target protein RNA, the level of CAT protein remained unchanged independently of light irradiation (Fig. 2). Similarly, the cell-cycle dependent promoter PrbcL did not exhibit any depletion in CAT protein accumulation throughout the entire length of the photoperiod. The constitutive promoter PdnaK was capable of stable and efficient production of the CAT protein. Probably the CAT protein is stable enough to remain in chloroplast at a level capable of ensuring sufficient protection from chloramphenicol through the 24-h long cell cycle. It might be caused by the particular stability of the exogenous CAT protein, lower protein turn-over cycle in the chloroplast or exceptionally high level of CAT synthesis in selected mutants. The single-cell colony cultures were grown in a step-wise increased concentration of chloramphenicol, thereby allowing for selection of high CAT expressers. The sequence optimization was also likely to contribute to greater RNA stability and rates of transcription and translation (Daniel et al. 2015). The stability of CAT protein was comparable with the stability of D1 protein (Fig. 2), which was established before as exceptionally stable (Krupnik et al. 2013) when purified to homogeneity. The chloroplast located expression of CAT protein was, most likely, most efficient in the stroma, what ascertained best conditions of pH for CAT to function (optimum in pH 7.6–8, with 50% activity at pH < 5 and > 9) (Thibault et al. 1980). This stability of the CAT protein had masked the modulatory property of inducible promoters (PpsbD and PrbcL); however, variable level of RNA transcribed for the respective genes was investigated before (Kanesaki et al. 2012). The length of the promotor sequences was relatively long, therefore other promoters located on the sequence might influence the strength of selected promoters. Also, the transcription might be driven by an entirely different locus-related, upstream promoter, resulting in a read-through polycistronic transcription. A sufficiently strong, the constitutive upstream promoter could drive polycistronic transcription and mask the modulatory effect of inducible promoters. It might be necessary to include a transcription terminator upstream of the inducible promoter to attain entirely inducible transcription of genes of interest.
We conclude that all the three studied promoters are suitable for promoting the transcription of the selectable gene (like cat) since they can ascertain very high and stable level of the resistance protein. The ability to achieve modulated protein expression (induced by light irradiation or in a specific phase of the cell cycle) is probably dependent on the specific location and read-though promoters, the inherent stability of that protein and / or its compatibility with the chloroplast proteolytic machinery of C. merolae.