tufA gene as molecular marker for freshwater Chlorophyceae
Article information
Abstract
Green microalgae from the class Chlorophyceae represent a major biodiversity component of eukaryotic algae in continental water. Identification and classification of this group through morphology is a hard task, since it may present cryptic species and phenotypic plasticity. Despite the increasing use of molecular methods for identification of microorganisms, no single standard barcode marker is yet established for this important group of green microalgae. Some available studies present results with a limited number of chlorophycean genera or using markers that require many different primers for different groups within the class. Thus, we aimed to find a single marker easily amplified and with wide coverage within Chlorophyceae using only one pair of primers. Here, we tested the universality of primers for different genes (tufA, ITS, rbcL, and UCP4) in 22 strains, comprising 18 different species from different orders of Chlorophyceae. The ITS primers sequenced only 3 strains and the UCP primer failed to amplify any strain. We tested two pairs of primers for rbcL and the best pair provided sequences for 10 strains whereas the second one provided sequences for only 7 strains. The pair of primers for the tufA gene presented good results for Chlorophyceae, successfully sequencing 21 strains and recovering the expected phylogeny relationships within the class. Thus, the tufA marker stands out as a good choice to be used as molecular marker for the class.
INTRODUCTION
The class Chlorophyceae comprises approximately 3,496 described species, according to Algaebase and is one of the most relevant phytoplankton groups in continental waters. The classification of this group is often hampered by the predominance of microscopic cells, frequently lacking obvious structures used to discriminate species or genera. Moreover, life habits, morphologic convergence favored by the unicellular form, the occurrence of cryptic species and asexual reproduction, which keeps mutations that can lead to a large morphologic variability (Potter et al. 1997) are factors that make the classification task arduous (Krienitz et al. 2001, Fawley et al. 2006, Krienitz and Bock 2012, Leliaert et al. 2012).
The urgency of a faster and practical classification system drives many investigations for an efficient molecular marker attending the premises of barcode concept from Consortium for the Barcode of Life (CBOL). This concept comprises the idea that molecular identifications should be conducted using a single pair of primers applicable in the most diverse groups of organisms, recovering a short marker (~700 bp) with enough variation for specific discrimination (CBOL Plant Working Group et al. 2009).
There are many markers proposed for different groups, such as the widely used cytochrome oxidase I (COX I), an official marker for some groups of animals, like fishes (Ward et al. 2005), red (Sherwood et al. 2008, Le Gall and Saunders 2010), and brown algae (McDevit and Saunders 2010), as well as diatoms (Evans et al. 2007).
In green algae, COX I is too variable requiring specific primers to be recovered in different taxa (Fučíková et al. 2011). The amplification of this gene has failed for some chlorophycean taxa (Hall et al. 2010). Furthermore, it may present introns (Turmel et al. 2002), hindering the design of new primers (Saunders and Kucera 2010).
Other markers are frequently used for phylogeny and identification studies of some algal groups, such as rbcL (rubisco large subunit), ITS (internal transcribe spacer), tufA (plastid elongation factor). Although widely used in phylogeny of green algae, 18S rDNA (Baldauf et al. 1990, Buchheim and Chapman 1991, An et al. 1999, Krienitz et al. 2001, 2002, Shoup and Lewis 2003, Hall et al. 2010, Buchheim et al. 2011) is a conserved gene (Luo et al. 2010, Fučíková et al. 2011) requiring other genes to solve closely phylogenetic relations in green algae. Moreover, many primers are necessary to recover it from different taxa (Garcia et al. in press), for example, used 12 primers to recover 18S rRNA gene from strains of one family within Chlorophyceae.
The most constructive results achieved so far have focused in phylogenetic questions for genera within the class (Van Hannen et al. 2000, Hall et al. 2010, Fučíková et al. 2011, McManus and Lewis 2011), therefore there is no known marker fulfilling the requirements of a universal barcode marker for Chlorophyceae.
Besides the universality, if the recovered marker has a good phylogenetic signal, it will allow a correct identification of a completely unknown organism, based on its phylogeny among others organisms already described. Thus, although unknown or undescribed, organisms can be classified in lower taxonomic levels if species discrimination is not possible, helping in culturing independent community studies, such as studies using massive sequencing platforms (Reyes et al. 2012, Salipante et al. 2013, Fumagalli et al. 2014).
According to the CBOL criteria of barcode applicability, the first step is to find primers that can recover those candidate molecular markers from the largest possible number of taxa. Thus, we aimed to test the universality of primers from published studies, already tested in other groups, for molecular markers in different orders of freshwater Chlorophyceae. Furthermore, we have built a phylogenetic tree with successfully sequenced marker, in order to investigate the possibilities of its application in the class.
MATERIALS AND METHODS
Strain cultures
All organisms are maintained in pure cultures in the Microalgae Collection at the Phycology Laboratory of the Federal University of São Carlos–Freshwater Microalgae Culture Collection (CCMA) (Portuguese acronym). Most strains were cultured in axenic conditions. The strains used in this study were classified and identified according to Algaebase sensu Komárek and Fott (Komárek and Fott 1983) (Table 1). Chaetophora sp. (CCMA-UFSCar 548) and Oedogonium sp. (CCMA-UFSCar 570) strains could not be classified further than genera. The only order from the Chlorophyceae that could not be tested was the Chaetopeltidales, due to the lack of isolates from this order in the culture collection.

Microalgae strains from CCMA.UFSCar collection and the amplification and sequencing results for each marker
Microalgae strains were cultivated in 100 mL Erlenmeyer flasks, with Wright’s Chryptophyte medium (Guillard and Lorenzen 1972), pH 7.0, 25 ± 1°C, light intensity of 300 µmol photons m-2 s-1 and a 12 : 12 light : dark cycle. Cultures in exponential growth phase, determined by optical density, were harvested in a centrifuge (Eppendorf 5415D; Eppendorf, Hamburg, Germany) under 3,500 ×g resulting in pellets of 40-60 mg of cells for DNA extraction.
DNA extraction and marker gene amplification
The concentrated material was homogenized by mixing in vortex for 15 seconds with glass beads (0.5 mm diameter) (Ningbo Utech International, Formosa, Taiwan) for mechanical cell disruption. The DNA was further extracted with Invisorb Spin Plant Mini Kit (Invitek, Hayward, CA, USA).
Strains of Nephrocytium lunatum and Pandorina morum form colonies with a thick polysaccharide envelope, which may avoid DNA extraction and hamper the polymerase chain reaction (PCR) reaction. For that reason, these strains were previously washed with lithium chloride to remove this envelope (Nordi et al. 2006).
Primers and PCR reaction
The primers tested for tufA, rbcL, and ITS (covering ITS1, 5.8S gene, and ITS2) markers, were chosen from published studies with organisms from class Chlorophyceae (Table 2). We tested two primers for rbcL gene, and their resulting fragments are overlapping each other. When both fragments were amplified from the same strain, they were submitted as a unique sequence with one access number.
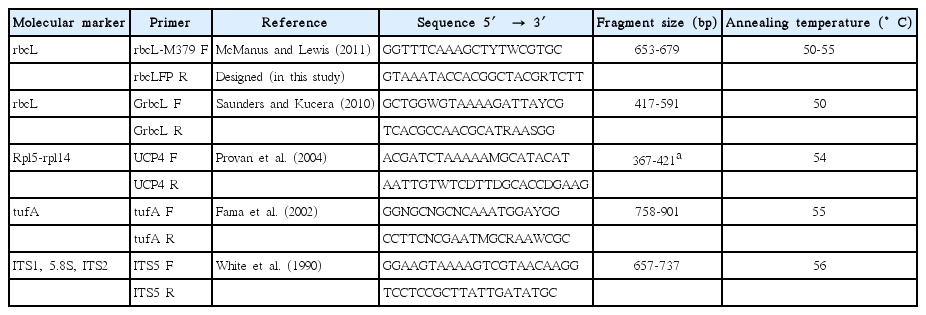
Molecular markers, names, and sequence of the tested primers
One of the pairs of primers tested for rbcL gene, rbcLFP, had the reverse primer designed in this study from sequences of Chlorophyceae available on the National Center for Biotechnology Information (NCBI). We also tested a pair of Universal Plastid Primers for Chlorophyta (UCP4) which recovers a portion of a plastidial gene, proposed by Provan et al. (2004).
The PCR mix was made as recommended by the Taq polymerase manufacturer (DNA polymerase, recombinant, 5 U µL-1; Invitrogen, Carlsbad, CA, USA) with 0.5 µM of each primer. The DNA was quantified by agarose gel electrophoresis using the ImageLab 4.0 (BioRad, Hercules, CA, USA) software and ranged from 5 to 10 ng.
PCR profiles were the same for all markers: initial denaturation for 4 min at 94°C; 29 cycles of 45 s at 94°C, annealing temperature specific for each pair of primers (Table 2) and 45 s of extension at 90°C followed by a final extension at 72°C for 7 min. Amplification was verified through electrophoresis in 1% agarose gel. In the case of amplification failure, changes in concentration of PCR reagents, DNA quantity and gradient of annealing temperature were tested, but none of these tests resulted in success of amplification (data not shown). PCR products were purified with polyethylene glycol 20% (polyethylene glycol) solution and NaCl 1 M (Lis and Schleif 1975) and the DNA sequencing was performed by Macrogen (Seoul, Korea).
Sequence analysis
Sequences were aligned with the CLUSTAL W software (Thompson et al. 1994) and the edition and protein frame reading translation, analysis of gaps, in/del and stop codons were performed at GENEIOUS version 6.1.7. Sequences were checked for contamination using the Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1990). Polymorphisms data, polymorphic sites, number of codons, synonym and non-synonym mutations, and parsimony informative sites were calculated with DNAsp 5.10 (Librado and Rozas 2009). Index of Substitution Saturation (ISS) and the Index of Substitution Saturations critic (ISSc) were calculated with the DAMBE5 v5.3.27 software (Xia et al. 2003) to evaluate if there was loss of phylogenetic signal by saturation of substitutions. Sequences were deposited in GenBank under the accession numbers found in Table 1.
Phylogenetic analysis
Phylogeny reconstruction was performed at Mr. Bayes (Huelsenbeck and Ronquist 2001) using a Monte Carlo Makov Chain (MCMC) with 3,000,000 generations, under the general-time-reversible nucleotide substitution model (GTR) (Rodríguez et al. 1990) including parameters for invariable sites (I) and gamma distributed rate variation (G), which was found using jModelTest v.0.1.1 (Darriba et al. 2012). Bootstraps values were obtained through neighbor-joining analysis, using 1,000 bootstrap replicates and genetic distances (p-distance) were calculated with MEGA 6 (Tamura et al. 2013).
For phylogenetic analysis with fragments of the tufA gene, sequences from GenBank were included to improve the representation of the order Chaetophorales (Schizomeris leibleinii UTEX LB 1228, accession number NC015645) and to represent the orders Chaetopeltidales (Floydiella terrestris UTEX 1709, accession number NC014346) which is lacking in our microalgae collection, and Oedogoniales (Oedogonium cardiacum UTEX 40, accession number EF587375), due to failure in sequencing the tufA gene of our representative strain. Furthermore, a sequence of Ostreococcus tauri (OTTH0595, accession number CR954199), class Mamiellophyceae, was included as outgroup.
RESULTS AND DISCUSSION
DNA amplification and sequencing
The tufA gene was easily amplified in all 22 strains. Only the strain Oedogonium sp. did not yield good sequences probably due to contamination, since this strain was not axenic (Table 1).
All the sequences obtained with tufA are new entries in GenBank, although there are tufA sequences deposited for the species K. aperta, P. duplex, and P. morum. The remaining 18 sequences which correspond to 15 species, since there are species with more than one strain, are new entries in the database for this marker.
After alignment of tufA sequences, gaps were not found and the final trimmed fragment had 743 bp, of which 305 were invariable sites, 438 were polymorphic sites displaying 716 mutations and 364 were parsimony informative sites. Amplified region was 247 codons, and the number of sites with synonym mutations was 172.26 and nonsynonym mutation was 568.74. Sequences set ISS value (0.32) was significantly lower (p = 0.001) than ISSc values (0.75 and 0.50) for symmetric and asymmetric trees, respectively, thus the phylogenetic signal was not hampered by the substitution saturation (Xia et al. 2003) also seen by (Fama et al. 2002, Fučíková et al. 2011).
Considering a lower taxonomic level, for example the family Selenastraceae which has more representatives (9 strains), the highest variation between two strains was 170 bases in a fragment of 826 bp (~20%), and the lowest variation was found between the three strains of the same species, Ankistrodesmus densus, 0-10 bases. Thus, the tufA marker was more variable than 18S rRNA gene for this family, since (Garcia et al. in press), for example, using 44 sequences of 18S rDNA (1,511 bp) of different genera of Selenastraceae, found the highest divergence of 76 bp. This higher variability, already shown in other studies of green algae (Hall et al. 2010), could make this gene more useful than the 18S rDNA for delimitation of lower taxonomic levels within the class.
The tufA gene codes for a molecule that mediates the entry of an amino-acyl-tRNA in the ribosome acceptor site during protein synthesis, dictating the peptide chain elongation to be formed. Due to its regulation function, it is a conserved gene (Delwiche et al. 1995), with intermediate evolution rate (Sáez et al. 2008).
The obtained fragment of the tufA gene is a partial coding sequence, being less vulnerable to major mutations that could have caused insertions, deletions or introns, which are unknown in green algae in this gene (Nozaki et al. 2002). Indeed, we have found no indications of introns, making this marker suitable to be tested as DNA barcoding for green algae, and appropriate for phylogenetic reconstruction.
The wide covering and sequencing success of the tufA gene with the primers tested here improves the results for the application of this marker in different groups, since it is already used for plasmodium, cyanobacteria and other bacteria, and terrestrial plants, with sequences available at the NCBI. This pair of primers has also been used in groups of macro (Du et al. 2014) and microalgae, such as cryptophytes (Garcia-Cuetos et al. 2010) and in the identification of microalgae present in the digestive tract of gastropods (Christa et al. 2013).
Furthermore, it has been widely applied in Ulvophyceae in different studies (Fama et al. 2002, O’Kelly et al. 2004, Wynne et al. 2009, Lawton et al. 2013) presenting great performance as DNA barcode for this class, except for the family Cladophoraceae (Saunders and Kucera 2010). In previous studies, species discrimination power of the tufA marker was observed for Ulvophycean (Fama et al. 2002, Saunders and Kucera 2010) and chlorophycean algae albeit they have used few genera from the class Chlorophyceae.
Although we have found that it is possible to recover tufA fragments from diverse chlorophycean taxa using a single pair of primers, the same could not be verified for the other markers tested (Supplementary Table S1).
For rbcL, it was not possible to perform the amplifications for all the strains using just one pair of primers. The GrbcL primers yielded good sequences for only 7 strains (Table 1), whereas the rbcLFP primers yielded 15 successful bidirectional sequences (Table 1). The rbcLFP primers had good performance from 50 to 55°C of annealing temperature (Table 2), although variations in the annealing temperature did not result in DNA amplification of the strains that failed to amplify in the first test.
A. densus (128) and Desmodesmus communis (030) yielded larger fragments (1,188 and 1,114 bp, respectively) than other strains when amplified with the rbcLFP primers. Comparing to a reference fragment from the NCBI, these larger sequences had an intermediate region (~800 bp) that could not be aligned with other sequences obtained.
This nucleotide sequence could correspond to an intron, what has already been reported for the rbcL in Chlorophyceae (Nozaki et al. 2002, McManus et al. 2012) (Supplementary Table S1). The presence of introns is not wanted in a candidate as a molecular marker since it hampers the design of primers and yields variable length fragments, complicating the sequence alignment. However, the nature of the intermediate portion can only be asserted through specific investigations, which were not the objective of this study.
The greater success of rbcLFP primers over GrbcL primers may be due to the fact that the first pair was specifically developed to be applied in class Chlorophyceae, using a forward primer chosen from a phylogenetic study with Pediastrum duplex (McManus and Lewis 2011) and a reverse primer designed in this study, from Chlorophyceae sequences available at GenBank.
The GrbcL primers were designed for application in Ulvophycean organisms (Saunders and Kucera 2010), in which authors tested different regions of the rbcL gene, finding better specific discrimination with the 3′ region, but more success of amplification with the 5′ region. Thus, the chosen pair of primers, aiming for universality, was the one that recovered the 5′ region.
However, the low amplification success and low quality sequences led to the exclusion of both rbcL primers as universal for class Chlorophyceae.
It must be noticed that although there is a large number of rbcL sequences available in GenBank for class Chlophyceae and other groups, they were often obtained using different primers and may be different regions of the gene, which makes their use as genetic markers for phylogeny or barcode less practical (Supplementary Table S1).
For the ITS region only 3 strains showed good sequencing (Table 1). The pair of primers ITS4-ITS5 for ITS region was chosen among proposed primers in a study with fungi phylogeny (White et al. 1990) and has already been tested with organisms from Chlorophyceae (Van Hannen et al. 2000, Buchheim et al. 2012).
Because it is a spacer region and is under a relaxed selection, mutations may not be strictly selected, which means it is very variable and may present in/dels and inconsistent sizes among the taxa, being commonly used for phylogeny within genus and species in green algae (Verbruggen et al. 2006, O’Kelly et al. 2010) (Supplementary Table S1). Thus, the highly variable nature of the ITS region may have contributed to its failure as a universal primer for Chlorophyceae, probably requiring particularly designed primers for each case.
Although the UCP4 primers have been proposed as universal for application in Chlorophyceae (Provan et al. 2004), no strain could be amplified following the protocol used in the original study, even when different annealing temperatures were tested. Provan et al. (2004) have tested the universality of primers for plastidial DNA using four organisms representing the Division Chlorophyta, with only one organism of the class Chlorophyceae, the specie Dunaliella salina.
The pair UCP4 was chosen in their study because the targeted region had the best combinations of characteristics for DNA Barcoding among the proposed regions, like constancy of non-coding sites number and the fragment size in the amplified lineages. Although the pair of UCP4 primers had worked for D. salina, it did not work for any of our strains.
Phylogeny
Concerning the use of the genes studied for phylogeny, the Bayesian tree topology with sequences of the tufA gene showed the monophyly of class Chlorophyceae and the five represented orders: Sphaeropleales, Chlamydomonadales, Oedogoniales, Chaetopeltidales, and Chaetophorales (Fig. 1).

Bayesian analysis for tufA sequences of Chlorophyceae. Sequences obtained in this study are indicated with “CCMA-UFSCar” and the four sequences obtained from GenBank are indicated with the name strain and the accession number. Ostreococcus tauri (Mamiellophyceae) was used as outgroup. Support values at the nodes are the bootstrap values (%) for neighbor-joining, followed by Bayesian posterior probability. Values lower than 95% for bootstrap value and 0.75 for Bayesian probability are represented by asterisk, or not presented when both were lower. The draws represent the basal body orientation of flagella apparatus, as the cells are represented as circles and seen from above. OCC, Oedogoniales Chaetopeltidales Chaetophorales; SC, Sphaeropleales Chlamydomonadales.
According to the flagella evolution (orientation of the basal body and number of flagella), it is possible to observe the Oedogoniales Chaetophorales Chaetopeltidales (OCC) clade, containing Oedogoniales, Chaetophorales, and Chaetopeltidales, and Sphaeropleales Chlamydomonadales (SC) clade, with Sphaeropleales and Chlamydomonadales. It is also in agreement with other studies that used the 18S rRNA gene (Alberghina et al. 2006, Němcová et al. 2011), 18S and 28S rRNA genes (Shoup and Lewis 2003) and nuclear and plastidial genes combined (Turmel et al. 2008, Tippery et al. 2012).
Despite not strongly supported (Bootstrap / Bayesian probability = 45/0.95), the monophyly of Sphaeropleales was shown with clear delineation of the families Selenastraceae and Scenedesmaceae (94 / 1.0 and 100 / 1.0, respectively) (Fig. 1). However, it is important to remember that some Sphaeropleales families were not represented here, and future works with the tufA gene must include them.
As many authors have already found using other genes (Fawley et al. 2006, Krienitz et al. 2011, Krienitz and Bock 2012), some internal branches were not clearly solved with superimposed genera, reflecting that genetic data may not behave consistently with morphology and leading to ambiguity in species delimitation. For example, the sickle morphology visible in Selenastraceae and used as identification also occurs in Trebouxiophyceae, indicating morphological convergence.
In summary, the tufA marker, standing alone, rebuilt the class Chlorophyceae phylogeny, which is often obtained with different genes combined, also at the internal branches, commonly addressed in specific investigations. Besides the overlap of some genera within Sphaeropleales, another issue that must be addressed is the position of N. lunatum. This species is currently classified as a Trebouxiophyceae member, but according to our phylogenetic reconstruction with the tufA marker, N. lunatum was positioned among Chlorophyceae, within the SC clade, close to Sphaeropleales and Chlamydomonadales (Fig. 1).
The Nephrocytium genus has already been classified in class Chlorophyceae, order Chlorococcales previously (West 1892, Pascher 1915), but families from this order were reorganized and redistributed. However, the transfer of the family to Trebouxiophyceae was based on the analysis of other genera (Friedl 1995) and the genus Nephrocytium was, apparently, passively transferred together with the other Chlorellaceae. Such taxonomic transferences have been already investigated, suggesting the resurrection of a Chlorophyceae genus to accommodate linages that were transferred to Trebouxiophyceae (Fučíková and Lewis 2012).
Nevertheless, the Nephrocytium genus is often missing in studies of phylogeny of Chlorophyceae and Trebouxiophyceae (Friedl 1995, Krienitz et al. 2002), and is underrepresented in this study, making a focused study with combined genes an essential procedure to elucidate its classification.
CONCLUSION
One of the critical characteristics for molecular markers is its applicability in as many organisms as possible. Among the 5 molecular markers tested here, tufA seems to comply with this objective for chlorophycean microalgae.
The easy amplification, sequencing and alignment of sequences, the crescent amount of available sequences on data bases summed with the good phylogenetic signal allowing a realistic phylogenetic reconstruction, despite the higher variability than 18S rRNA gene, indicate the tufA gene as a promising molecular marker for the class. However, its utilization as a DNA barcode in Chlorophyceae, alone or combined with others markers, need to be tested in further studies, comprising problematic taxa, such as family Selenastraceae.
Despite the rbcL primers not amplifying all the strains they could be applied for green algae genera / species in focused studies. The primers tested for ITS and UCP4 regions were not appropriate for universal application in Chlorophyceae due to their low amplification / sequencing success rate.
Supplementary Materials
Advantages and disadvantages of each molecular marker tested in this study and other principal markers used in studies with green algae (http://e-algae.org).
Acknowledgements
We would like to thank Thaís Garcia da Silva for the morphological identification of the microalgae strains. We also wish to thank Dr. Pedro Manoel Galetti Junior for the suggestions made for the development of this work.
Abbreviations
BLAST
Basic Local alignment Search Tool
CBOL
Consortium for the Barcode of Life
CCMA
Freshwater Microalgae Culture Collection (in Portuguese acronymic)
COXI
cytochrome oxidase I
GTR
general-time-reversible nucleotide substitution model
ISS
Index of Substitution Saturation
ISSc
Index of Substitution Saturations critic
ITS
internal transcribed spacer
MCMC
Monte Carlo Makov Chain
NCBI
National Center for Biotechnology Information
OCC
Oedogoniales Chaetopeltidales Chaetophorales
PCR
polymerase chain reaction
rbcL
large unit ribulose bisphosphate carboxylase (gene)
SC
Sphaeropleales Chlamydomonadales
tufA
elongation factor tu (gene)
UCP
universal chlorophyte primers