Evolutionary history of the monospecific Compsopogon genus (Compsopogonales, Rhodophyta)
Article information
Abstract
Compsopogon specimens collected in China were examined based on morphology and DNA sequences. Five molecular markers from different genome compartments including rbcL, COI, 18S rDNA, psbA, and UPA were identified and used to construct a phylogenetic relationship. Phylogenetic analyses indicated that two different morphological types from China clustered into an independent clade with Compsopogon specimens when compared to other global samples. The Compsopogon clade exhibited robust support values, revealing the affiliation of the samples to Compsopogon caeruleus. Although the samples were distributed in a close geographical area, unexpected sequence divergences between the Chinese samples implied that they were introduced by different dispersal events and from varied origins. It was speculated that Compsopogon originated in North America, a portion of the Laurentia landmass situated in the Rodinia supercontinent at approximately 573.89–1,701.50 million years ago during the Proterozoic era. Although Compsopogon had evolved for a rather long time, genetic conservation had limited its variability and rate of evolution, resulting in the current monospecific global distribution. Additional global specimens and sequence information were required to increase our understanding of the evolutionary history of this ancient red algal lineage.
INTRODUCTION
Compsopogon Montagne 1846 is a typical Rhodophyta algal genus that inhabits freshwater and is globally distributed (Kumanoa 2002). This genus can be morphologically identified by observing the C. caeruleus morphology with regular polyhedral cortical cells or the C. leptoclados morphology with irregular cortical cells and rhizoidal outgrowths (Necchi et al. 2013). Taxonomic characteristics within the genus Compsopogon are debatable and have been substantially debated. Systematic features of the species delineations in this genus include the type of the basal portion of the thallus, the branching pattern and the number of cortical layers. However, these characteristics are widely variable both within and among populations and with different environmental factors (Necchi et al. 1999). Krishnamurthy proposed a new genus Compsopogonopsis V. Krishnamurthy 1962, which belonged to the order Compsopogonales (Krishnamurthy 1962). The species numbers in these two genera were variable according to different studies and sampling zones (Krishnamurthy 1962, Necchi and Ribeiro 1992, Vis et al. 1992). However, Necchi (Necchi et al. 2013) examined 25 specimens of Compsopogon across a wide geographical distribution (excluding Asia) and proposed that these two genera were synonymous and only one species was identified, Compsopogon caeruleus (Balbis ex C. Agardh) Montagne, supporting the previous proposal of the investigation by Rintoul of Compsopogon in North America (Rintoul et al. 1999). Compsopogon is globally distributed, but primarily in tropical and subtropical zones, with a few reports in temperate regions (Sheath and Hambrook 1990). In China, the populations of this genus are scarce, and only four species of Compsopogon and one species of Compsopogonopsis have been reported in previous literature (Shi 2006), whereas these species probably represent C. caeruleus according to the new taxonomic proposal of genus Compsopogon.
Molecular biological methods have important application potential for investigating all systematic levels of Rhodophyta (Freshwater et al. 1994, Vis et al. 2007, 2012, Entwisle et al. 2009, Necchi et al. 2013, Salomaki et al. 2014). The results of these molecular biological techniques have produced different taxonomic assignments contradicting traditional morphological observations, notably for the genus Compsopogon. Molecular phylogeny suggested that C. caeruleus, the sole species recognized in genus Compsopogon has a worldwide distribution with considerable morphological plasticity but little genetic variation (Necchi et al. 2013). Therefore, all other previously identified species were considered synonymous to C. caeruleus (Necchi et al. 2013). However, limited molecular data were previously available for Compsopogon. The majority of the sequence data originated from specimens in North America, South America, and Australia, whereas molecular data for this genus are almost absent from Asia, except for one specimen in Japan (Necchi et al. 2013).
As an early derived lineage within Rhodophyta (Freshwater et al. 1994, Yoon et al. 2004, Yang et al. 2016), Compsopogon is critical for investigating the phylogenetic position within the red algae and in providing insights into the evolution process of this ancient algal lineage. An understanding of the evolutionary history of this ancient emerging algal genus will benefit investigations in paleogeography and paleoclimatology and of the evolution of our planet. The molecular investigation of Chinese Compsopogon is important to preserve and enrich the genetic diversity of the available information. Although diverse investigations into the divergence times and historical biogeography of higher-class plants based on molecular analyses have been reported recently (Xie et al. 2009, Deng et al. 2015), no related reports have covered members of the order Compsopogonales. Therefore, we have undertaken a primary investigation of the ancestral geographical distribution and origin of the genus Compsopogon to understand the evolutionary process of this important taxonomic unit.
This study was performed to determine molecular data of this genus sampled in China, to analyze the genetic variability of specimens and to determine whether its taxonomic assignment was consistent with the proposal of a single species in this genus. We compared the phylogenetic relationships established by analyzing DNA sequences of different genes including molecular markers for the nucleus, chloroplast, and mitochondria. Additionally, we inferred the geographic origin of this genus based on the modern distribution pattern and the divergence time of this algal lineage.
MATERIALS AND METHODS
Sample collection, pretreatment, and morphological observations
Two Compsopogon specimens, MM09010 and MM13006, were collected from Shanxi Province, China at different times and locations (Table 1). The samples were cleaned with distilled water. Epiphytes were then removed under a dissecting microscope. A portion of the samples were stored at −20°C prior to DNA extraction. The remaining sample portions were soaked in a formalin solution for the morphological observations. Both the features of the entire thalli and the cell characteristics were examined, and the images were recorded with a camera (DP72; Olympus, Tokyo, Japan) attached to a microscope (BX-51; Olympus).
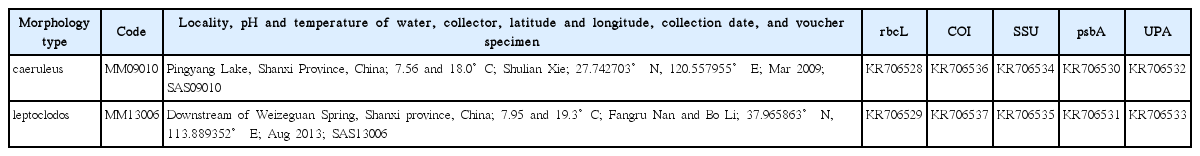
Morphology type, collection information, GenBank accession numbers for specimens in this study
DNA amplification and sequencing
Samples were ground in liquid nitrogen, and the total DNA was then extracted following the protocol described by Saunders (1993) with modifications following Vis and Sheath (1997). Sequences covering the nuclear small subunit rDNA (SSU), plastidial ribulose-1,5-bisphosphate carboxylase-oxygenase large-subunit gene (rbcL), photosystem II reaction center protein D1 (psbA), 23S ribosomal RNA gene (UPA), and mitochondrial cytochrome c oxidase subunit I (COI) were polymerase chain reaction (PCR) amplified with previously described specific primers (Supplementary Table S1). Specific primers for c18s5 and c18s3 were designed to amplify the SSU fragment of MM09010 because the previously reported general primers did not amplify the sequence. PCR amplifications were conducted in 20 μL volumes containing 12.5 μL ddH2O, 2.0 μL 10× buffer, 2.0 μL 2.5 mM dNTPs, 0.2 μL Taq DNA polymerase (all from Sangon, Shanghai, China), 2.0 μL of primer (10 mM), and 1.0 μL of genomic DNA. Typical thermal cycling conditions for rbcL included an initial denaturation step at 95°C for 2 min, 35 cycles at 93°C for 1 min, 47°C for 1 min, 72°C for 2 min, and a final extension at 72°C for 2 min. For COI, the conditions for the run were an initial denaturation step at 94°C for 4 min, 35 cycles at 94°C for 1 min, 45°C for 30 s, 72°C for 1 min, and a final extension at 72°C for 7 min. For the SSU, the conditions for the run were an initial denaturation step at 94°C for 4 min, 38 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 1.5 min, and a final extension at 72°C for 7 min. For psbA, the conditions for the run were an initial denaturation step at 94°C for 2 min, 35 cycles at 94°C for 30 s, 46.5°C for 30 s, 72°C for 1 min, and a final extension at 72°C for 7 min. For UPA, the conditions for the run were an initial denaturation step at 94°C for 2 min, 35 cycles at 94°C for 20 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 10 min.
The PCR products were purified using a SanPrep column DNA gel purification kit (Sangon) and then were sent to BGI Tech Corporation (Beijing, China) for sequencing on an ABI 3730XL sequencer (Applied Biosystems, Foster City, CA, USA). Both amplification primers were used to determine the sequences. Sequences were inspected manually with Sequencher ver. 4.14 (Codes 2000). The DNA sequence data generated from this study have been deposited in GenBank (the accession numbers are listed in Table 1).
Sequence alignment and phylogenetic analysis
The sequences obtained in this study and related sequence data of freshwater Rhodophyta downloaded from GenBank (listed in Supplementary Table S2) were assembled in Clustal-X 2.0 (Thompson et al. 1997). Untrimmed bases on both ends were deleted to produce an identical length alignment. The sequence characteristics were calculated as a complete data matrix in MEGA 5.0 and both the total base pairs and variable base pairs of each sequence were calculated (Tamura et al. 2011). Specimens of Compsopogon were divided priori into groups according to their distribution area (including groups originating from Asia, Europe, Australia, South America, North America, and the Pacific), and sequence variations among and between groups were analyzed in MEGA 5.0. The gene sequences were then used to reconstruct phylogenetic trees. Appropriate evolutional models were inferred using Modeltest ver. 3.7 (Posada and Buckley 2004), with the results listed in Supplementary Table S3. The Neighbor-joining method was performed in the MEGA 5.0 (Tamura et al. 2011). The Kimura 2-parameter model was selected as the substitution model, and the bootstrap repetition was set to 1,000. Both transitions and transversions were considered. Maximum likelihood trees were built using PHYML software (Felsenstein 1981, Guindon and Gascuel 2003). The bootstrap analysis was conducted using 1,000 replicates. Additionally, Bayesian inferences were developed in MrBayes ver. 3.1.2 (Ronquist and Huelsenbeck 2003). A Markov chain Monte Carlo (MCMC) was initiated in the Bayesian inference and run for 5,000,000 generations; the trees were sampled every 1,000 generations. A consensus tree was summarized after 1,000 trees of burn-in. Independent and combined sequences with available information including rbcL, COI, and SSU were loaded into the software SplitsTree to reticulate the network relationship. The neighbor-net method was selected and numbers of bootstrap replicates were set to 1,000 (Huson and Bryant 2006). The genus Cryptomonas belonging to Cryptophyta was designated as the outgroup (according to an earlier investigation of red algal) (Ragan et al. 1994).
Estimation of divergence time
Estimation chronograms were constructed for all of the three independent sequences (rbcL, SSU, and COI) using Beast v1.7.5 (Drummond and Rambaut 2007). Sequences of UPA and psbA were not used to construct dated tree because the serious scarcity of sequence information in GenBank database, which will severely limit the analysis. BEAUti software was used to set up the files to run Beast. Time-calibrated trees were constructed using the Bayesian approach in Beast v1.7.5. Taxa were divided into four groups based on the phylogenetic analysis: ingroup, outgroup, Bangiophyceae and Florideophyceae, with monophyly constraints on the outgroup, ingroup and Florideophyceae. The analysis was performed using the general time reversible (GTR) nucleotide substitution model (determined by Modeltest in the phylogenetic analysis) with a Gamma distribution for four rate categories. To account for the uncertainty in the divergence time estimation, an uncorrelated lognormal relaxed clock model was employed (Drummond et al. 2006). The time of divergence and the credible intervals were calculated using the Yule pure birth model of speciation and the tree model for the random starting tree. Posterior distributions of parameters were approximated after 50,000,000 generations of MCMC runs, sampling every 5,000 generations with a 10% burn-in. The convergence of the chains was determined using the program Tracer v 1.6 (Rambaut et al. 2014).
Fossil calibrations and estimated divergence time information were used for the relaxed molecular clock model to infer the divergence time. The fossil calibration used in this study was derived from well-preserved abundant thalli in the Doushantuo Formation which were affiliated to the modern red algal class Florideophyceae and dated back to 570 ± 20 million years ago (MYA) (Xiao et al. 1998). Doushantuo algal thalli are characterized by branching forms with tissue differentiation and specialized reproductive structures similar to the carposporangia and spermatangia of living red algae (such as the genus Batrachospermum in the class Florideophyceae). Consequently, this calibration was selected as constraint for the monophyletic group of Florideophyceae. Additionally, with genus Cryptomonas functioning as the outgroup, we set the treeModel rootHeight to 1,274 ± 5 MYA, which was estimated as the splitting time for Cryptophyta (Yoon et al. 2004). The priors on the age of the nodes were set as a normal distribution with means and standard deviations that were consistent with the fossil ages or estimated ages.
Ancestral geographical origin of the genus Compsopogon inferred using RASP
Based on the tree data and the final tree generated in Beast, we reconstructed the ancestral geographical origin of the genus Compsopogon using a Statistical Dispersal-Vicariance Analysis implemented in the RASP (Reconstruct Ancestral State in Phylogenies) software (Yu et al. 2010, 2011). Only dated trees based on rbcL and COI sequences were used to reconstruct the ancestral geography. Distribution ranges for Compsopogon were divided into six areas: A, Australia; B, South America; C, North America; D, Asia; E, Europe; and F, the Pacific. To account for the phylogenetic uncertainty, we used 10,000 trees from the Beast output files and discarded the first 1,000 trees as the burn-in. At 850–1,000 MYA, Rodinia was separated into two halves by the Panthalassic Ocean, forming the northern landmass constituted of the China-Australia-Antarctic continents and the equatorial flange landmass constituted of North America and South America (Li et al. 2008). Therefore, the maximum areas of each node was set to 3 (A, D, and E or B, C, and F) and the geographical combinations was set in accordance with this definition. All other options remained as default. Because we mainly focus on the ancestral geographical origin of the genus Compsopogon, the other unrelated clades, including the Florideophyceae clade and the outgroup clade, were not shown in result.
RESULTS
Morphological characterization
The MM09010 specimen displayed a bolder and darker colored thallus with nearly regular cortical cells, whereas the MM13006 specimen displayed a thinner and lighter colored thallus with irregular cortical cells characterized by rhizoidal cells emerging from the cortical cells (as illustrated with the arrow in Fig. 1).
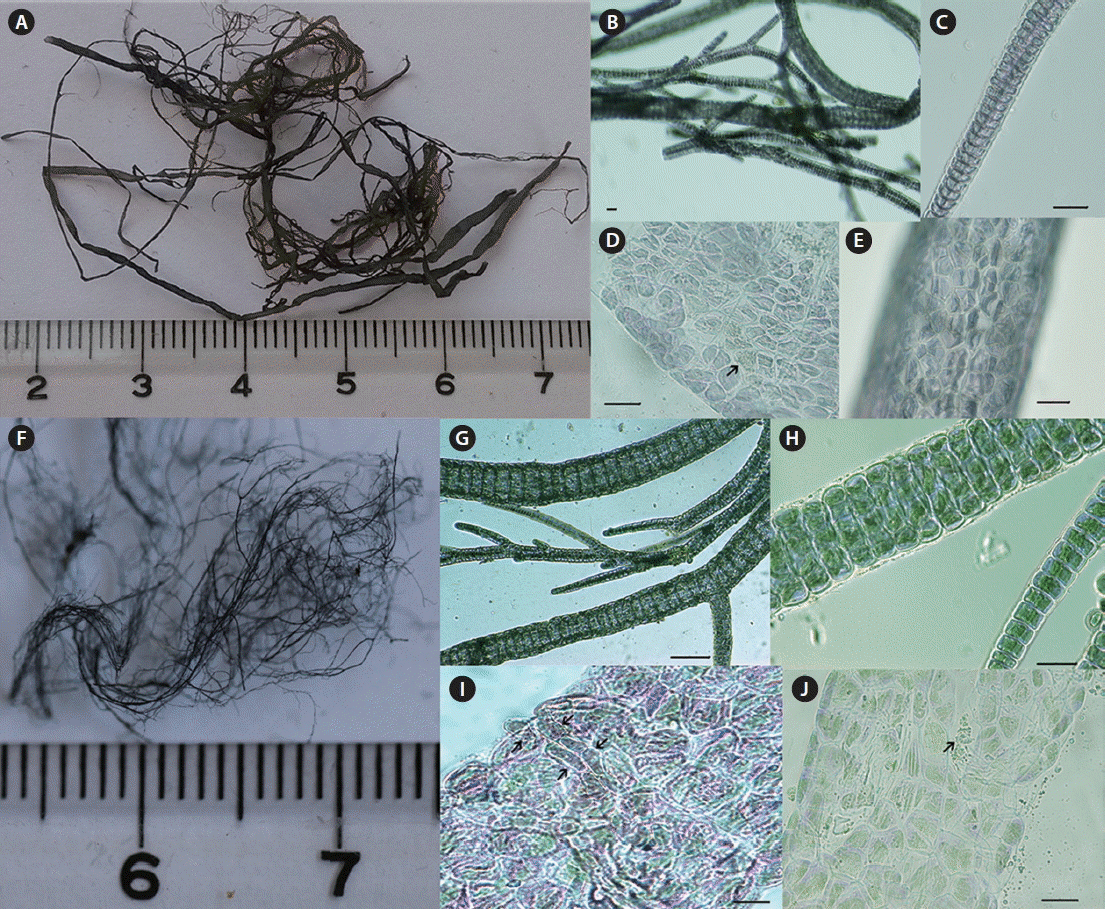
Morphology characteristics of the specimens in this study. (A) Morphology of the entire thallus for MM09010. (B) Multiple branches of the thallus. (C) Immature thallus with a single-layer cortical cell. (D & E) Surface view of the cortical cell arranged regularly; the arrow refers to monosporangia. (F) Morphology of the entire thallus for MM13006. (G) Multiple branches of the thallus. (H) Developing thallus with single-layer cortication for the central axis formation. (I) Surface view single-layer cortication; the arrows refer to rhizoidal cells. (J) Surface view of the cortical cell arranged irregularly; the arrow refers to microsporangia. Scale bars represent: 50 μm.
Sequence characteristics
Within all the sequences, sequence length of UPA was the shortest. The COI covered the largest percent of parsimony informative sites whereas the UPA showed the greatest value for the transition and transversion ratio. The sequence variance rate was the lowest for the nuclear SSU marker and the highest for the mitochondria COI marker (Table 2). The pairwise distance analysis between and within the geographical groups for genus Compsopogon showed that within group distances for rbcL and COI in North America were higher than the between group distances of North America and other geographical continents (Table 3), whereas the within group distance for the nuclear SSU in North America was lower than that of the Pacific group. For Asian samples, rbcL sequence between the specimens from Japan and China (MM13006) were identical, whereas the two Chinese specimens shared distances of 0.0014 and 0.0006 for rbcL and SSU sequences, respectively (Table 4).

Characteristics of the sequences used in this study

Distance matrixes between and within groups for the genus Compsopogon based on the rbcL, COI, and SSU

rbcL and SSU sequence distance of Asian specimens in this study
Phylogeny revealed by different molecular markers
All methods (Bayesian inference, maximum likelihood, and neighbor-joining) used in analyzing independent and combined rbcL, SSU, and COI sequences generated similar tree topologies and only the Bayesian tree of the combined sequences was illustrated in Fig. 2A and all other independent phylogenetic trees were shown as supplementary figures (Supplementary Figs S1–S3). According to the consistent tree topology, Compsopogon samples in this study clustered together with other specimens of this genus forming a Compsopogon clade (Fig. 2A) with robust supporting values in all analyses. The morphotypes from China, MM09010 and MM13006, respectively, formed a sister group with samples from other continents. Compsopogonophyceae and Florideophyceae used in the phylogenetic reconstruction formed monophyletic branches, and were supported robustly. All Compsopogon specimens were clustered into a linear split in the Neighbor-net phylogram, divergent from the Florideophyceae taxa at an early time and shared no horizontal gene communication (Fig. 2B).

Phylogenetic analysis phylogram based on combined gene sequences. For Bayesian inference tree, the support values for all analyses are shown as follows: Bayesian posterior probabilities / maximum likelihood bootstrap / neighbor joining distance bootstrap. ‘-’ denotes <50% support for that analyses at that node. For the Neighbor-net phylogram, the numbers marked on the branches denote the support values in percent after 1,000 bootstraps. (A) Bayesian inference tree based on combined sequences. (B) Neighbor-net phylogram based on combined sequences.
Based on UPA and psbA sequences, all Compsopogon specimens with available sequences clustered into a main branch representing the Compsopogonophyceae clade; this clustering was supported robustly in all three methods (Supplementary Figs S4 & S5). With no other sequence information of Compsopogon specimens, MM09010 and MM13006 formed an independent terminal branch in the psbA phylogeny.
Divergence time estimation
Based on the time inference using the rbcL sequence (Fig. 3A), the divergence time of genus Compsopogon was 917.02 MYA (95% highest posterior density [HPD]: 573.89–1,701.50) (Fig. 3A). The COI sequence estimated the divergence time of the genus Compsopogon as 859.23 MYA (95% HPD: 578.32–1,023.56) (Fig. 3B). However, the nuclear SSU sequence estimated the split of Compsopogon occurring at 763.20 MYA (95% HPD: 326.78–1,301.67) (Supplementary Fig. S6), significantly later than the results based on the rbcL and COI sequences. The results based on the rbcL and COI sequences were largely consistent in predicting the Compsopogon divergence during the Proterozoic Era. Estimation on the time of Compsopogon species introduced into China was not statistically solved, neither the time of other dispersal events to different distribution areas.
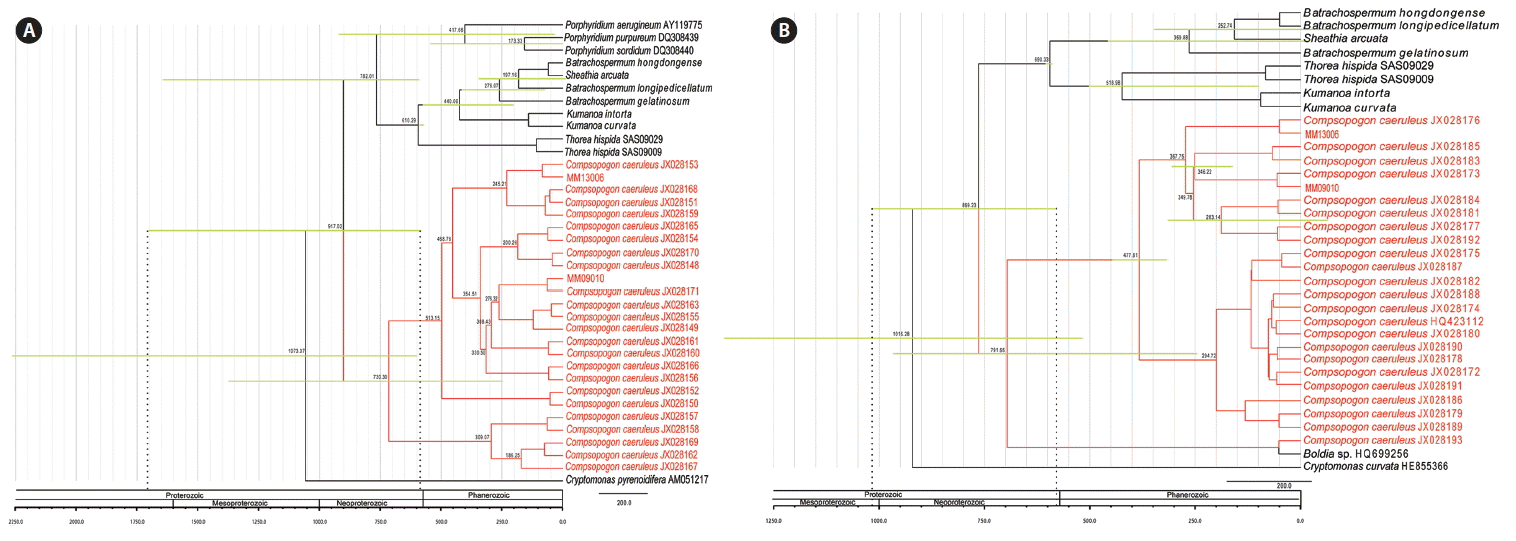
Divergence time estimation phylogram based on different gene sequences using BEAST. Numbers at the nodes represent the mean estimated divergence time with the corresponding 95% highest posterior density of the important nodes indicated by horizontal bars. The geological timescale is given in million years ago. Asterisks denote the calibration points. The taxa in red in the estimated tree represent the genus Compsopogon. (A) Divergence time estimation tree based on the rbcL sequence. (B) Divergence time estimation tree based on the COI sequence. rbcL, ribulose-1,5-bisphosphate carboxylase-oxygenase large subunit; COI, cytochrome c oxidase subunit I.
Ancestral geographical origin inference
Both geographical reconstruction trees (rbcL and COI) displayed a similar origin area (Fig. 4). The geographical origin of the genus Compsopogon was traced back to the American plate, more likely in the North American area, as shown in node 63 (combination of South America and North America, relative probability = 0.46) for rbcL (Fig. 4A) and node 66 (North America, relative probability = 0.56) for COI (Fig. 4B). This adjacent area was referred to as “Laurentia” and was located in the southern part of the “Rodinia” supercontinent at approximately 850–1,000 MYA. After the originating in “Laurentia,” the genus presumably dispersed to North America and South America, respectively with the separation of these two continents. Both MM09010 and MM13006 were introduced into China from the American plate but through different dispersal events.
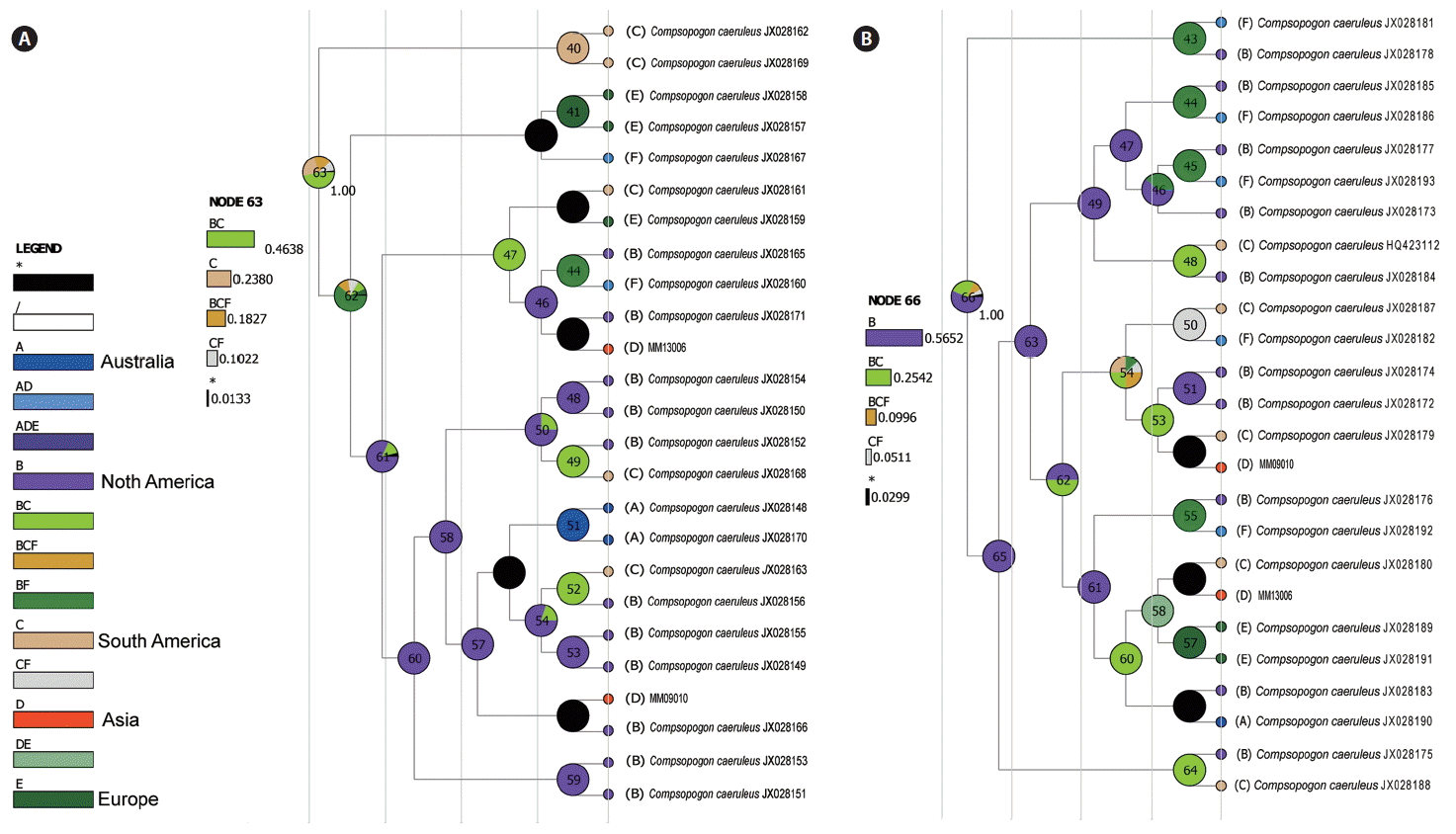
Ancestral area reconstruction using the S-DIVA based on different gene sequences. The current distribution area of accessions is provided at the tips of the branches. (A) Origin reconstruction based on the rbcL sequence. (B) Origin reconstruction based on the COI sequence. rbcL, ribulose- 1,5-bisphosphate carboxylase-oxygenase large subunit; COI, cytochrome c oxidase subunit I.
DISCUSSION
According to traditional taxonomic assignments, the Chinese specimens in this study represented different morphological types of Compsopogon with both the caeruleus MM09010 and the leptoclados morphology MM13006, as treated by Necchi et al. (2013). The clustering of these two specimens into the Compsopogon clade with high supporting values implied the synonymy of the genera Compsopogon and Compsopogonopsis (Seto 1987, Xie and Ling 2003), which was consistent with a previous proposal (Shyam and Sarma 1980, Rintoul et al. 1999, Necchi et al. 2013). Additionally, the highly supported Compsopogon clade suggested that the Compsopogon specimens from China presently analyzed were indeed C. caeruleus.
The Neighbor-net phylogram displayed a linear relationship, suggesting the early divergence of genus Compsopogon from the Florideophyceae taxa and the minor sequence variance during their independent evolutionary process. The intraspecific pairwise distances of genus Compsopogon were 0–0.23, 0–0.55, and 0–0.06% for rbcL, COI, and SSU, respectively. Intraspecific variation of rbcL and COI sequence in other red algae had been reported as 1.2–7.2 and 0–6.5%, respectively and the nuclear SSU was more conserved (Freshwater and Rueness 1994, Vis et al. 2010). Little genetic variation with large geographic distances was also observed in other freshwater Rhodophyta and the explanations were assumed to recent dispersal events or genetic bottlenecking (House et al. 2010, Rueness 2010). Given the antiquity of the red algae, we speculated the reason leading to low genetic variation of genus Compsopogon was genetic bottlenecking, which limited its genetic variation during the long evolutionary history. The worldwide distribution and low genetic variation of genus Compsopogon were previously inferred to be caused by its asexual reproduction (Necchi et al. 2013). The reproduction pattern combined with its strict habitat choice, which occurred only in freshwater and distributed primarily in tropical and subtropical areas with a few in temperate regions, resulted in the current single species constitution of genus Compsopogon. Additionally, genetic divergences between Chinese specimens though collected in close geographical areas were noted in this study. One potential explanation for this divergence was that they originated from different haplotypes.
The divergence time estimation based on organelle markers including rbcL and COI obtained similar results. Both analyses estimated the split of Compsopogon to occur in the Proterozoic era (95% HPD: 573.89–1,701.50 MYA). In Hunting Formation reports, fossils of exceptional cellular preservation have allowed a detailed resolution of the taxonomy attached to the Bangiophyceae dating back to 1,174–1,222 MYA (Butterfield 2001). Yoon et al. (2004) used the plastid multi-gene markers to infer the molecular timeline for the origin of photosynthetic eukaryotes and estimated the split of the red and green algae to have occurred 1,474 MYA. The origin time of Compsopogon obtained in our study was consistent with the time range for red algae divergence based on fossil evidence and molecular-based calculation results. Assuming that the estimation of our study was reasonable, the emergence of Compsopogon occurred in the Neoproterozoic-Mesoproterozoic, during which algal radiation was observed in fossil records and in other molecular clock analyses (Yoon et al. 2004). The mitochondrial COI marker, sharing common feature of uniparental inheritance with the chloroplast genome, revealed consistent divergence time estimation with the chloroplast gene-based results. However, the nuclear SSU was reported to be more conserved when compared with the chloroplast and mitochondrial molecular sequences (Pareek et al. 2010). In other molecular phylogeny investigations of Rhodophyta, the SSU was shown to be not practical for species discrimination because of the high degree of conservation (Müller et al. 2001). We speculate that the high conservation of nuclear SSU make it not an appropriate molecular marker for divergence time estimation in ancient genus Compsopogon.
The reconstruction of the geographical area of origin for this genus suggested that its speciation occurred on the American plate, most likely in North America. As was observed in this study, Compsopogon specimens in North America exhibited more sequence diversity than other geographical groups. Approximately 1,100 MYA, the east coast of North America was adjacent to western South America, both forming the Laurentia landmass (Park 1992). At the time of origin for Compsopogon, the Laurentia landmass was located in the equatorial flange. Therefore, it is speculated that this tropical climate in American plant triggered the speciation of this genus. In the subsequent years, the genus Compsopogon dispersed with the rifting and recombination of the continents. Probably due to the habitation in water, this genus survived the mass species extinction that occurred at the end of the Permian (Benton and Twitchett 2003, Schulte et al. 2010). Globally, the climate during the Cretaceous Period, was much warmer than today, and warm water from equatorial regions migrated northward. These factors benefited the dispersal and vicariance of Compsopogon across the Tethys to other continents (Pearson et al. 2001).
Rhodophyta formed a main lineage in the first endosymbiosis event. This event is of great value in paleobiology investigations and studies of the evolution process of the earth climate and geography. This study infers the evolutionary history of the anciently derived red algae Compsopogon. With further specimens and sequence information sampled across the globe, the derivation and development of red algae will be better understood.
SUPPLEMENTARY MATERIAL
Supplementary Table S1. Specific primer sequences of different molecular markers used in this study (http://www.e-algae.org).
Supplementary Table S2. Specimen information of sequences downloaded from the GenBank database (http://www.e-algae.org).
Supplementary Table S3. Substitution models obtained for each gene sequence using Modeltest 3.7 analysis (http://www.e-algae.org).
Supplementary Fig. S1. Phylogenetic tree based on the rbcL sequences (http://www.e-algae.org).
Supplementary Fig. S2. Phylogenetic tree based on the COI sequences (http://www.e-algae.org).
Supplementary Fig. S3. Phylogenetic tree based on the SSU sequences (http://www.e-algae.org).
Supplementary Fig. S4. Phylogenetic tree based on the UPA sequences (http://www.e-algae.org).
Supplementary Fig. S5. Phylogenetic tree based on the psbA sequences (http://www.e-algae.org).
Supplementary Fig. S6. Divergence time estimation tree based on the SSU sequences (http://www.e-algae.org).
ACKNOWLEDGEMENTS
This study was funded by the National Natural Science Foundation of China (No. 31370239 and 31670208 to Shulian Xie). We sincerely appreciate the English language editing of Nature Publishing Group Language Editing.