INTRODUCTION
Diatoms are eukaryotic unicellular microalgae with unique cell walls (frustules) made of silica. Synthesis of new biosilica cell wall occurs in a special membrane-bound intracellular compartment called silica deposition vesicle (SDV). Though the immediate precursor for silica formation inside the SDV is unclear, monosilicic acid [Si(OH)4], which occurs in natural habitats, clearly represents the original source for silica formation. Silicic acid freely diffuses across the cell membrane at concentrations greater than 30 μM (Thamatrakoln and Hildebrand 2008). Although the average concentration of oceanic silicic acid is around 70 μM, diatoms still need an efficient uptake system. This is because the level of silicic acid is generally less than 10 μM in surface waters where the diatoms usually inhabit (Tréguer et al. 1995, Martin-Jézéquel et al. 2000) and the intracellular levels of silicic acid can be several thousand-fold higher than the environmental concentration in some cases (Martin-Jézéquel et al. 2000). Therefore, diatoms require the transporters for silicic acid to pump this acid into cells and possibly into the SDV, to overcome this difference of concentration. This transporter also appears to play a critical role in regulating the levels of intracellular pools of unpolymerized soluble Si, thus preventing polymerization prior to deposition in SDV (Hildebrand 2000).
Silicon transporters (SITs) were first identified in the marine pennate diatom, Cylindrotheca fusiformis, as a novel family of transporters with no known homologs (Hildebrand et al. 1998). These membrane-associated proteins have 10 transmembrane domains and can directly interact with and transport silicic acid. Multiple hybridizing signals in Southern analyses and data from whole genome sequencing and de novo assembly of transcriptome indicate that SITs are encoded by a multigene family in diatoms (Hildebrand et al. 1998, Sapriel et al. 2009, Durkin et al. 2016). The genome of Fragilariopsis cylindrus contains seven SIT genes belonging to four different clades (A to D) (Durkin et al. 2016). Hildebrand et al. (1998) hypothesized that the SIT proteins encoded by multigene family have unique roles in the uptake of silicic acid according to their subcellular locations, Si binding affinities, and transport rates. Also, the possible regulatory role has been suggested for some Thalassiosira pseudonana SITs (TpSITs), in which they sense if the silicic acid levels are sufficient to proceed with cell wall formation and cell division (Shrestha and Hildebrand 2015).
The synchrony of the unicellular culture has been demonstrated especially invaluable for the study of molecular, biochemical, and cellular processes closely coupled to the cell cycle (Hildebrand et al. 2007); synchronization can increase the sensitivity in detecting the physiological or molecular changes coupled to the cell cycle. Diatom biosilica cell walls have a structure like a petri dish, with one half (epitheca) overlapping the other half (hypotheca). Each theca is composed of valve and girdle bands, which are synthesized in separate specialized membrane-bound intracellular compartments termed SDV. Synthesis of a new girdle band appears to facilitate cell expansion, while new valve establishment allows cell division (Hildebrand 2008).
Molecular genetic mining for the key factors in biosilicification in new species can provide new insights for the understanding of this process. For example, it allows comparative studies with already identified members, facilitates the discovery of new factors that are possibly related to the unique structural characteristics of the species, and can provide information of the diverse species-specific regulatory circuits of the process which have been evolutionarily established for the adaptation to the specific environment. F. cylindrus is the one of the most successful psychrophilic diatoms in the Southern Ocean, serving as the primary source of nutrition and energy for this ecosystem (Sackett et al. 2013). Also, it has a relatively small genome (approximately 60 megabase pairs), whose sequence has been recently determined (Mock et al. 2017). Genomic datasets for F. cylindrus are available in the web portal of the Joint Genome Institute (JGI; http://genome.jgi.doe.gov/Fracy1/Fracy1.home.html).
In this study, we have established a synchronized culture of F. cylindrus by depletion and repletion of light or silicate, and examined the timing of syntheses of girdle bands and valve, and expression of F. cylindrus SIT genes (FcSITs) during the progression of cell cycle.
MATERIALS AND METHODS
Algal strain and growth experiments
The Antarctic marine diatom, F. cylindrus strain (CCMP 1102) was obtained from the National Center for Marine Algae and Microbiota (East Boothbay, ME, USA). This strain was grown in artificial seawater medium (f medium) (Guillard and Ryther 1962) supplemented with biotin and vitamin B12 of 4.1 × 10−9 and 7.38 × 10−10 M, respectively. Cultures were maintained at 4°C on orbital shaker at 100 rpm under continuous illumination (30 μmol photons m−2 s−1). For growth analysis, 20 mL of f medium or medium depleted of specific nutrient was inoculated with exponentially growing cells at a cell density of 1 × 10−6 cells mL−1 and cultured as described above. For growth analysis, the cell density of each culture at the indicated times was determined by microscopic counting using a hematocytometer (Marienfeld Superior, Baden-Württemberg, Germany). The results are presented as the mean ± standard deviation.
Synchronization procedure
Cultures were inoculated with exponentially growing cells (1 × 10−6 cells mL−1) and grown as described above. At the exponential stage of growth (5 × 106 cells mL−1), the cells were either transferred to dark conditions or silicate-free [Si(−)] medium. For the inoculation into Si(−) medium, the cells were washed twice with Si(−) medium by centrifuging at 3,500 rpm 4°C for 5 min in a Combi-514R centrifuge (Hanil Science Industrial, Incheon, Korea) and resuspended in Si(−) medium. This suspension was then used as inoculum for the Si(−) culture. Culture in normal f medium in the dark and in Si(−) medium with continuous illumination were maintained at 4°C with shaking as described above. After 24 h, the dark-arrested cells were shifted to continuous illumination and further cultured up to 30 h. Si starvation-arrested cells were transferred to the complete f medium by removing all the medium by pelleting the cells at 3,500 rpm 4°C for 5 min in the aforementioned centrifuge, resuspending the cells in normal f medium, and culturing up to 30 h. In the experiment where synchrony was induced by deprivation of both silicate and light, exponentially growing cells were transferred to the Si(−) medium as described above and cultured in the dark for 24 h. The resulting cells with an arrested cell cycle were transferred to fresh complete medium and incubated in the presence of Si, light, or both.
RNA extraction
To isolate total RNA for the study of gene expression, 50 mL of culture containing approximately 5 × 106 cells mL−1 was obtained from cultures during growth under the specific conditions at indicated times. The cells were recovered by centrifuging at 3,500 rpm and 4°C for 5 min in the aforementioned centrifuge. All procedures for RNA extraction were carried out according to the manufacturer’s instructions for RNA iso Plus (Takara Bio, Otsu, Japan).
Quantitative real-time RT-PCR (RT-qPCR)
Total RNA was used for synthesis of first-strand cDNA using an iScript cDNA Synthesis kit (Bio-Rad, Richmond, CA, USA) following the manufacturer’s instructions. RT-qPCR using a Thermal Cycler Dice Real Time System (Takara Shuzo, Kyoto, Japan) and SYBR Green (Takara Bio) was carried out as described by Park et al. (2010). RT-qPCR cycling comprised a 30 s pre-denaturing step at 95°C followed by 45 cycles of a 5-s denaturing step at 95°C, a 10-s annealing step at 55°C, and a 20-s extension step at 72°C. The relative amplification of the gene encoding the TATA-box binding protein (TBP) was examined as an internal control to normalize all data. Triplicates of each sample were examined to evaluate the quantitative variation in each sample, and each experiment was repeated at least three times. The gene-specific primers used for RT-qPCR are listed in Table 1.
Fluorescence imaging of new cell walls
HCK-123 (LysoTracker Yellow HCK-123; Thermo Fisher Scientific, Waltham, MA, USA) was used to label the newly synthesized cell wall. HCK-123 (1 mM stock solution in dimethyl sulfoxide) was added to the medium at a final concentration of 1 μM when the cell cycle was re-started by resupplying the limiting factor. At the indicated times, a small volume of culture was removed and the cells were observed for the presence of fluorescently labeled new cell wall using fluorescence microscopy with an Axioskop microscope (Carl Zeiss, Gottingen, Germany) equipped with an X-Cite 120 fluorescence microscope illumination system (Lumen Dynamics, Mississauga, ON, Canada). Digital images were captured with an AxioCam MRm microscope camera (Carl Zeiss) using AxioVision Rel 4.8 software (Carl Zeiss Microimaging Inc., Jena, Germany).
Flow cytometry analysis
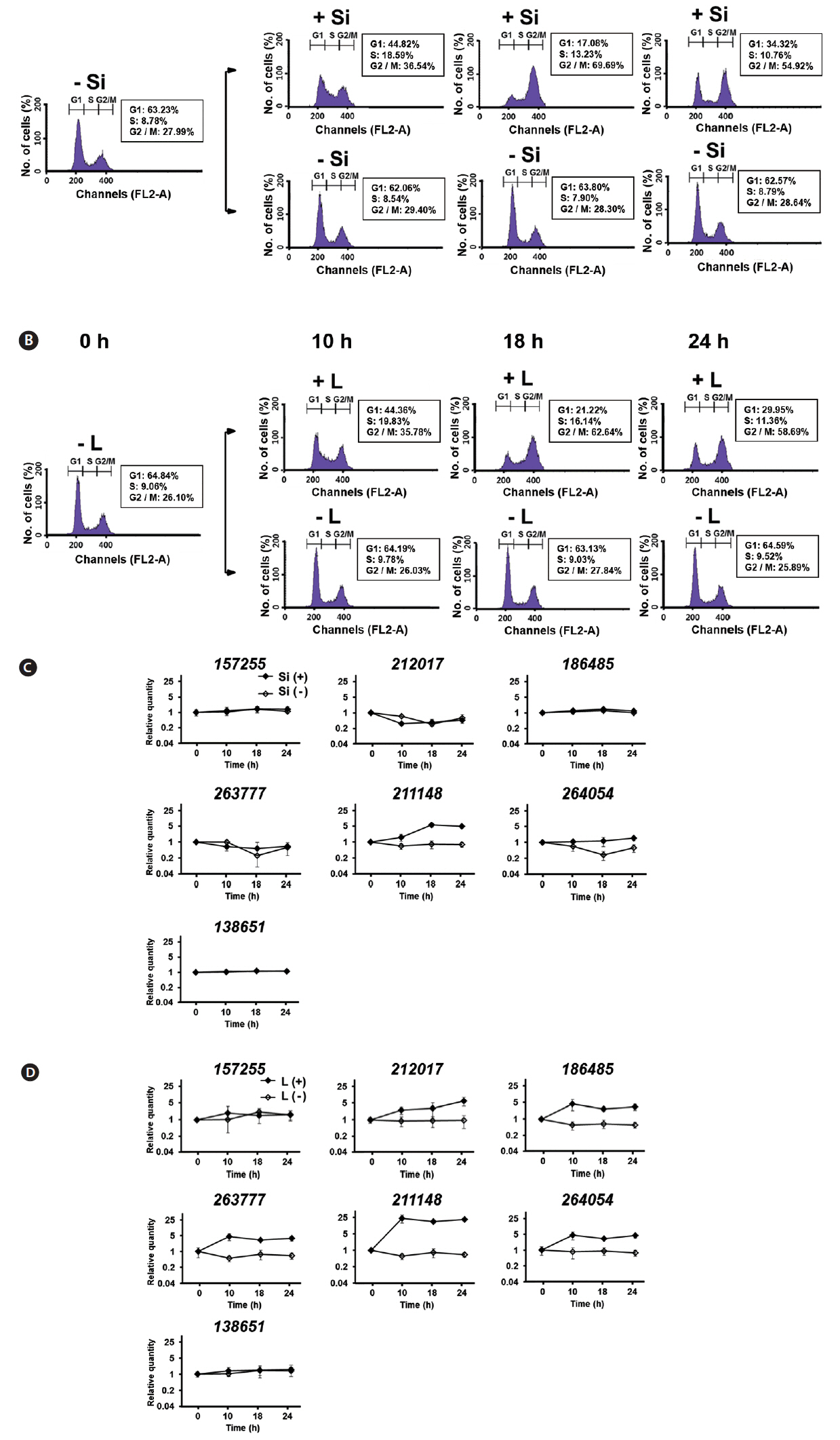
Flow cytometry for the analysis of the cell-cycle stage was performed using a FACS Calibur apparatus (Becton-Dickinson Corp., Franklin Lakes, NJ, USA). For this analysis, cells were recovered by a brief period of centrifugation and resuspended in 600 μL phosphate buffered saline (PBS) buffer (20 mM potassium phosphate monobasic buffer, pH 7.4) at a cell density of 1 × 107 cells mL−1. The cells were fixed with ethanol by mixing with 1,400 μL of ethanol for at least 1 h. The fixed cells were washed once with PBS buffer, resuspended in PBS buffer containing propidium iodide (PI, 50 μg mL−1) and DNase-free RNase A (100 μg mL−1), and incubated for 30 min in the dark. The PI-stained cells were washed once with PBS buffer and resuspended in 500 μL of PBS buffer. The stage of the cell cycle was determined by monitoring the amount of PI fluorescence from the cell DNA. The relative percentage of cells in the G1, S, and G2/M phases was determined using CellQuest Pro software (Becton-Dickinson Corp.) as described by Hildebrand et al. (2007).
RESULTS AND DISCUSSION
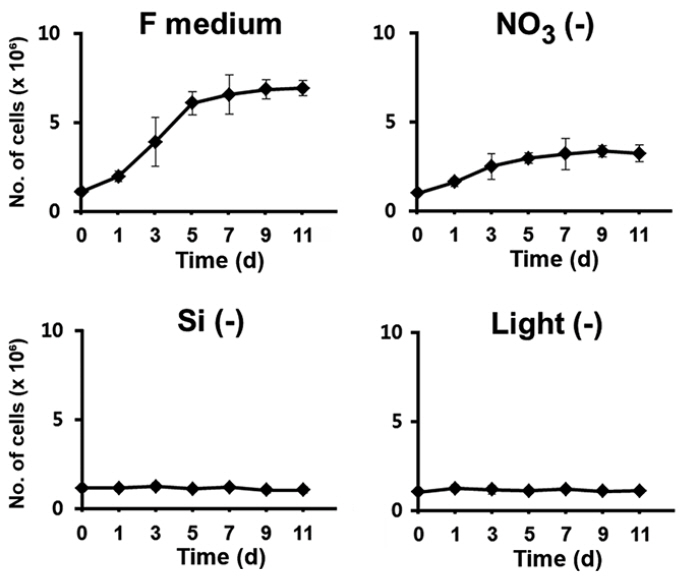
Growth of Fragilariopsis cylindrus culture is effectively restricted by depletion of silicate or light
We first examined the factors that were most effective in restricting the growth of F. cylindrus cultures upon depletion, since arrest of cell growth by depletion of a specific factor can be released by its repletion, allowing the subsequent synchronous progression of the cell cycle. To identify the key factors, exponentially growing cells were transferred to the normal complete culture medium in the absence of light or to a continuously illuminated medium depleted of a specific nutrient such as nitrogen and silicon, which are the major limiting nutrients of primary production in the ocean environment (Falkowski et al. 1998). The growth was determined in each culture after the transfer (Fig. 1). When F. cylindrus cells were maintained in the normal medium under continuous light, the typical sigmoidal pattern of growth was evident, with more than a 6-fold increase in cell number in less than 7 d. Nitrogen can be considered indispensable for diatom growth because very large amounts of it are required as a major constituent for the synthesis of amino acids and nucleic acids (Valenzuela et al. 2012, Yang et al. 2013). Indeed, it has been demonstrated that nitrogen limitation in diatoms prolongs the duration of the G1 phase or arrest the diatoms at multiple G1 checkpoints (Olson et al. 1986, Vaulot et al. 1987). However, the growth was limited only up to 50% of that of the control in the absence of nitrate. The sustained growth of the culture to a certain level even in the absence of nitrate may be because the cells utilize the nitrogen stored intracellularly in nitrate-free conditions, resulting in delayed arrest of cell cycle.
In contrast, deprivation of either Si or light rapidly and tightly restricted the growth of F. cylindrus culture, suggesting that either of those factors was entirely required for the cell division process. The newly formed silica cell wall separates two daughter cells, and completes the cell division process. Since new silica cell wall cannot be established without a silicate supply, it is expected that a silicate deficiency would arrest the growth of cell culture. The availability of Si is a recognized major limiting factor for diatom reproduction in the ocean environment (Falkowski et al. 1998). Light is one of the most important factors that influence the growth for photosynthetic organisms, including diatoms. Diatoms appear to have evolved specific photoacclimation and photoadaptation mechanisms, which makes them able to survive over a wide range of light intensities and wavelengths (Huisman et al. 2004, Lavaud et al. 2004, 2007, Schellenberger et al. 2013). Nonetheless, their cell division seems to be highly light-dependent; the light-entrained cell division of diatoms indicates that the cell cycle has light controlled restriction points. In our examination, the growth of F. cylindrus culture was also immediately restricted by the absence of light.
Fragilariopsis cylindrus cell cycle can be synchronized by deprivation and repletion of Si or light
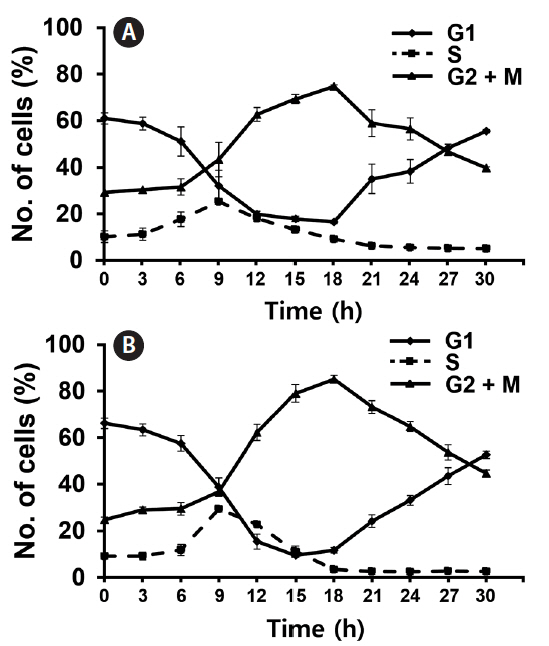
Since both Si and light deprivation could effectively prevent the growth of F. cylindrus culture, we examined if the cultured cells would remain in particular phases of the cell cycle when these factors were limited. Re-addition of the depleted factor to the cells arrested at a specific stage of the cell cycle allows the culture to synchronously progress through the cell cycle. The relative amounts of F. cylindrus cells at the specific stages of cell cycle after deprivation of either silicate or light for 24 h and following replenishment of the depleted factor were monitored by staining DNA with PI and measuring the fluorescence in individual cells by flow cytometry (Fig. 2). To synchronize cell division in F. cylindrus cells, the exponentially growing cells were subject to darkness or Si-starvation, to arrest the growth and released synchronously by illumination or Si-repletion. Fig. 2A and B present the synchronized progression through the cell cycle of F. cylindrus cells by depletion and repletion of either silicate or light. Si-starvation successfully arrested the majority of the cell population at G1 phase in F. cylindrus. However, the stage of arrest by Si-starvation appears to vary depending on the diatom species. For example, T. pseudonana arrests predominantly in G1 phase (Hildebrand et al. 2007), Thalassiosira weissflogii in both the G1 and G2 phases (Brzezinski et al. 1990), and C. fusiformis at the G1/S phase boundary (Darley and Volcani 1969). We also tested the effect of the prolonged period of starvation (2-d), it did not improve the arrest efficiency in our examination. As shown in Fig. 2A, the majority of F. cylindrus cells (63%) starved for Si for 1 d were arrested at the G1 phase, 8% in S phase, and 29% in the G2/M phase. The peak of the S phase occurred at 9 h and maximum G2/M at 18 h. After about 36 h after Si repletion, similar levels of the cells had returned to the G1 phase, suggesting the synchronized progression through the cell cycle (data not shown). As shown in Fig. 2B, 67% of F. cylindrus cells were arrested at the G1 phase after 1 d in light-deprived conditions, with 10 and 23% arrested in the S and G2/M phase, respectively. The peak of the S phase occurred at 9–10 h and the maximum G2/M arrest was at 18 h. Similar to silicate, the second peak of the G1 phase was predicted to occur approximately 36 h after re-irradiation in the similar levels of the F. cylindrus culture. A similar light-induced synchronization method has been proven useful to synchronize the cell division of Sandalolitha robusta (Gillard et al. 2008) and Phaeodactylum tricornutum (Huysman et al. 2010). Accordingly, light limitation and deprivation experiments have identified light-controlled restriction points in several diatom species, either only during the G1 phase or during both the G1 and G2/M phases of the cell cycle (Olson et al. 1986, Vaulot et al. 1986, Brzezinski et al. 1990, Gillard et al. 2008, Huysman et al. 2010).
The total length of the F. cylindrus cell cycle was approximately 36–48 h in our examination. This is much longer than the 4 to 6 h that was evident for T. pseudonana. For example, T. pseudonana cells peaked at S phase 3 to 4 h after the restart of the cell cycle and the G2 phase peak was reached after 4 to 6 h (Hildebrand et al. 2007). In case of F. cylindrus cells, maximum DNA synthesis (S phase) occurred at 9 h following the restoration of either silicate or light and the majority of cells reached the G2/M peak at 18 h. This is well matched with the previous report that F. cylindrus showed an approximately 3 times lower exponential growth rate under optimal growth conditions than T. pseudonana (Bender et al. 2014).
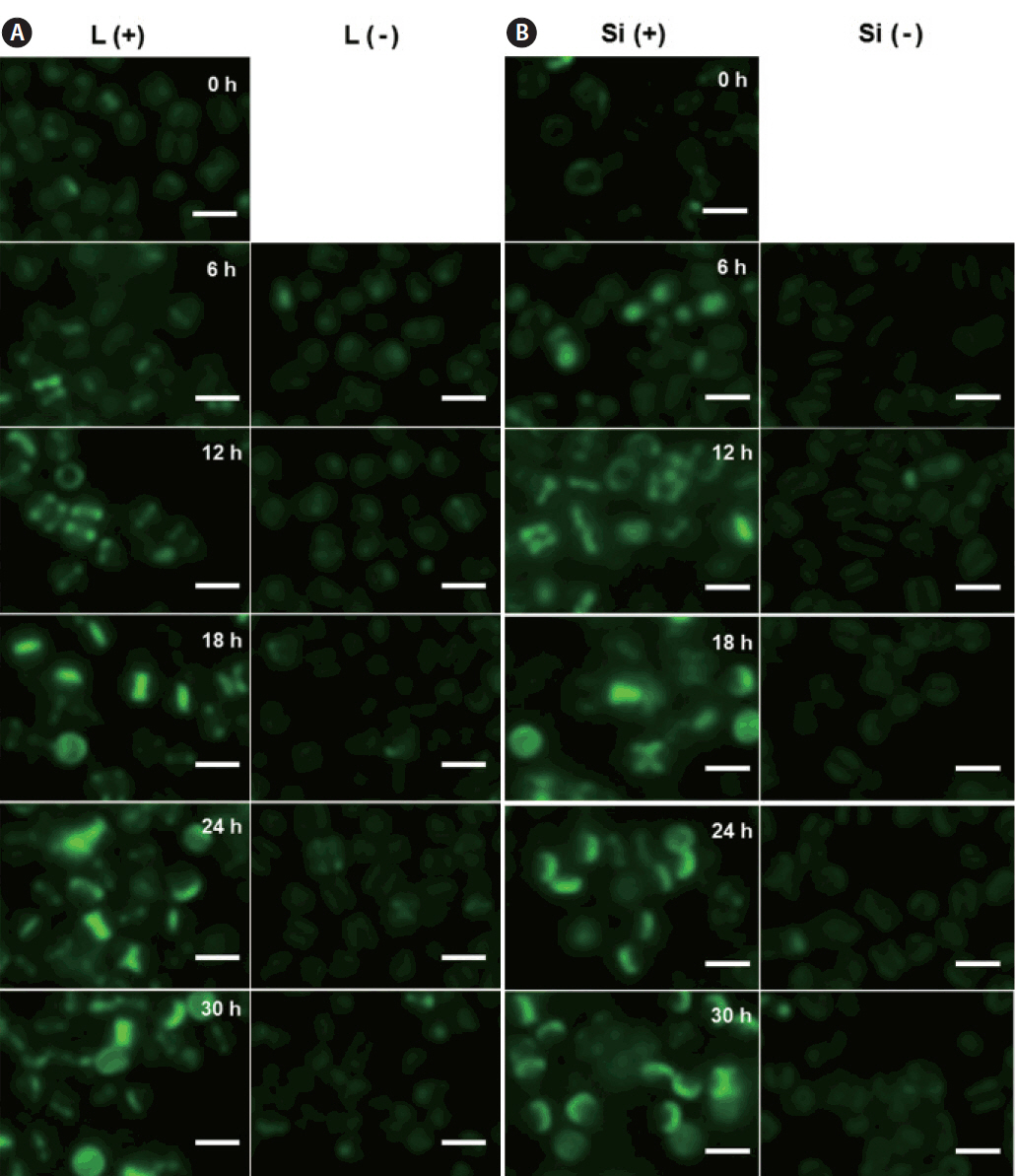
Fluorescence staining analysis of new cell wall synthesis
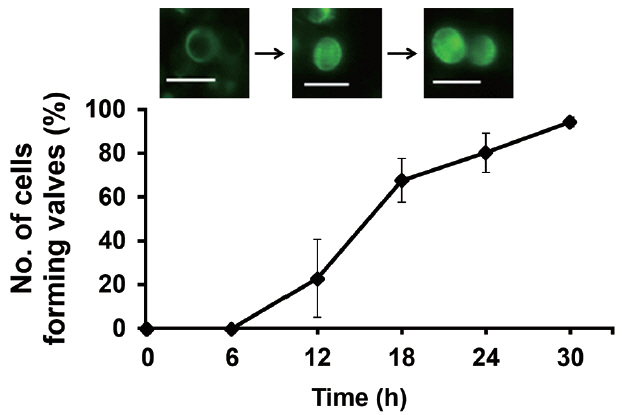
We investigated the dynamics of frustule formation during cell cycle. Diatom frustules have been successfully imaged using few fluorescent tracers including rhodamine-123 (Li et al. 1989, Brzezinski and Conley 1994, Hildebrand et al. 2007), the LysoSensor Yellow/Blue DND-160 (Shimizu et al. 2001, Hazelaar et al. 2005, Vrieling et al. 2005, Frigeri et al. 2006). In our study, biosilica structures newly established was examined by HCK-123, an acidotropic fluorescent dye that is stable, allowing successive acquisitions and higher signal-to-noise ratio, compared to other dyes (Descles et al. 2008). When F. cylindrus cells were released from cell cycle arrest by Si repletion or illumination, a marginal amount of very faint labeling of the girdle bands was detectable at 6 h. The majority of the cells showed strong fluorescence at around 18 h, where the cell cycle would be at the peak of the G2/M phase (Fig. 3A & B). Fig. 4 shows the relative amounts of cells with new girdle bands that established new valves during the cell cycle of Si-synchronized cultures. No cells with new girdle bands had developed new valves approximately 6 h after the re-initiation of the cell cycle, indicating that the initiation of the girdle band synthesis occurred first, followed by valve formation. About 20% of cells displayed valve synthesis until S-phase, while more than 70% of cells with new girdle bands showed new valves at 18 h after the re-initiation of the cell cycle. This suggests that most active valve synthesis appears to take place during the G2/M phase.
Differential expression of FcSIT genes during the synchrony by Si or light
Since the functional diversification is the important characteristics of the multigene family, expression profiles of FcSIT multigenes were accessed using differently synchronized cultures. In our study, expression of seven FcSITs was examined at three indicated times in Si- or light-synchronized cells in which the cell cycle had reached the S- or G2/M-phases and returned to the G1 phase. The timing of cell cycle progression of every culture used for the study of FcSIT expression was confirmed by flow cytometry (Fig. 5A & B). Previously, Durkin et al. (2016) classified the diatom SIT genes into five distinct clades (A through E) through the phylogenetic analysis and suggested their possible functional divergence. Interestingly, we found that expression patterns of FcSIT multigenes show the clade-specific pattern of expression. A significant induction was observed for one FcSIT gene (JGI protein ID 211148) with no induction in other FcSIT genes in Si-synchronized cells, upon restoration of Si (Fig. 5C). Si has been shown to repress the expression of SIT genes in various diatoms, including T. pseudonana, P. tricornutum, and Pseudo-nitzschia multiseries (Sapriel et al. 2009, Shrestha and Hildebrand 2015, Brembu et al. 2017). Expression of some SIT genes of F. cylindrus was also negatively affected by the presence of silicate (Durkin et al. 2016). For example, all the FcSIT genes belonging to clade A (212017, 263777, and 264054) were highly upregulated upon Si-starvation. We think that the lack of responsiveness to Si in those clade A genes in Fig. 5C is due to the discrepancy in the experimental design. In our experiment, the transcript abundance through the cell cycle is normalized to the levels measured during Si-starvation, which would have already induced upregulation of these SIT A genes. Therefore, it is not surprising that those SIT A genes show no change over the cell cycle when synchronized by Si because they were already induced by a stress response. Actually, we also confirmed the induction of expression of those genes by Si-starvation (Supplementary Fig. S1). In light-synchronized cells, expression of all FcSIT genes except two genes (157255 and 138651) were increased during the progression of cell cycle (Fig. 5D). In this light-synchronized conditions, those SIT A genes with clades C (186485) and D (211148) are all upregulated during the cell cycle, suggesting that they may play a certain role in Si uptake during the cell cycle under replete conditions. Durkin et al. (2016) also hypothesized that the FcSITs in clade B (157255 and 138651) are related to a more universal Si metabolism present in non-diatoms, so it is not surprising that these 2 SITs are not differentially transcribed during cell cycle progression at any conditions.
Previously, the expression of TpSIT1 and TpSIT2 was shown to peak at the S-phase of synchronized cultures (Thamatrakoln and Hildebrand 2007). RNase protection assays using Si-synchronized C. fusiformis cultures indicated that all of the SIT genes (SIT1 to SIT5) are expressed most strongly just prior to the period of maximum Si incorporation in the cell wall synthesis (Hildebrand et al. 1998). A rapid increase up to the S phase and a sustenance pattern was observed in the expression profiling of FcSIT genes. There was an alteration in gene expression during the synchrony of the cell cycle by either silicate or light, rather than a peak-form of expression. However, we cannot still exclude the possibility that relatively sparse number of samplings might miss a peak-form of expression too.
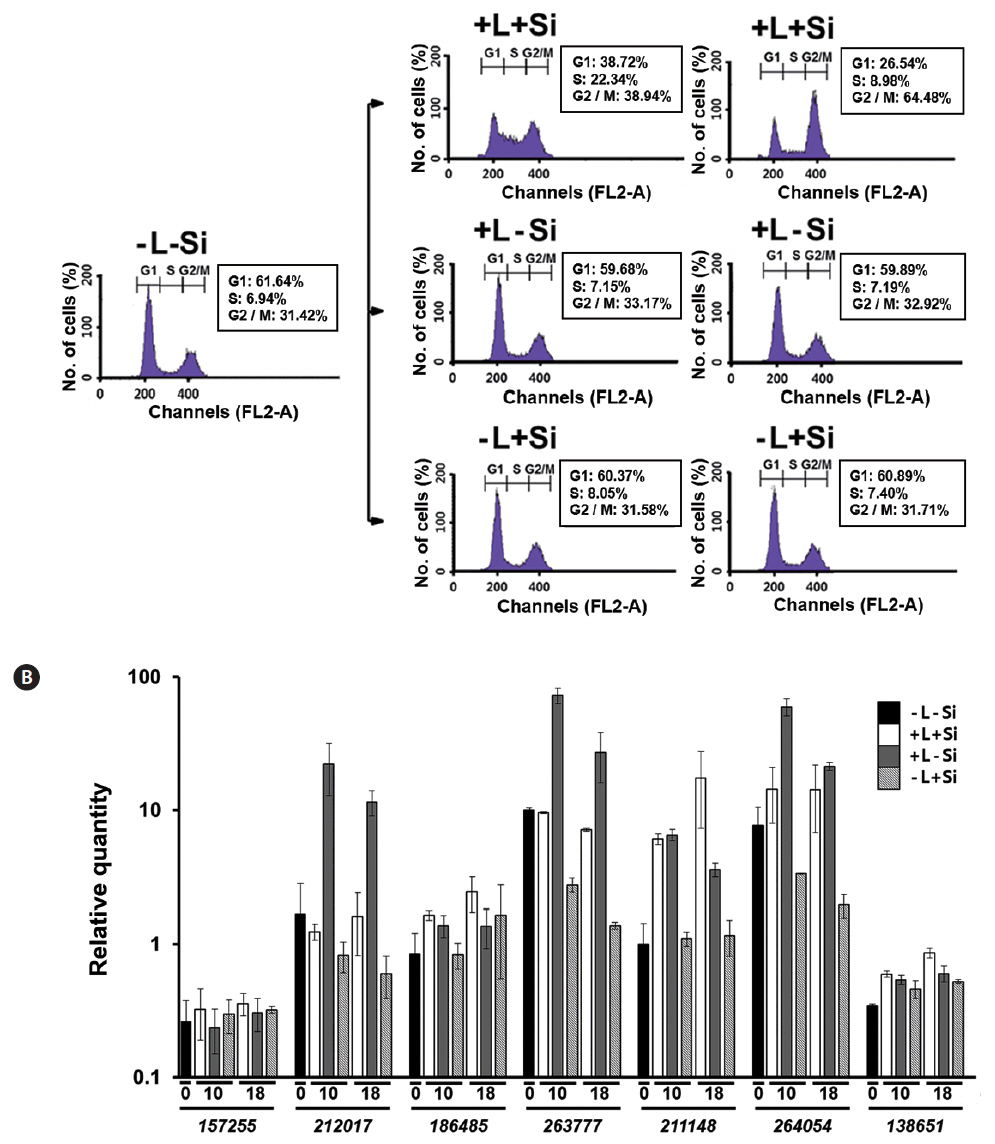
To examine if the altered expression of FcSIT genes is tightly coupled to the progression of the cell cycle, we monitored the expression pattern of FcSIT genes in Fc cultures that had been simultaneously synchronized by both Si and light. For example, G1-phase-arrested Fc cells were prepared by transferring exponentially growing cells to the silicate-depleted medium and maintaining in darkness for 24 h. The cell cycle of this culture could be re-started only with the repletion of both light and silicate, while remaining arrested at the G1 phase when repletion involved only one of the factors (Fig. 6A). As shown in Fig. 6B, the abundance of those clade A FcSIT transcripts (212017, 263777, and 264054) was decreased with time in cells cultured with Si in the absence of light, compared to those in cells at time 0 (−L-Si). Also, the levels of those transcripts were much higher in cells cultured without Si in the presence of light, than those cultured with Si and light. These results are consistent with the fact that Si suppresses the expression of those genes. Also, expression of the FcSIT genes belonging to clades A and C that were found to be light-inducible (Fig. 5D) displayed good induction in response to light in Fc cells that remained arrested at the G1 phase due to the depletion of Si. Therefore, the progression of cell cycle itself is unlikely required for light induction of those FcSIT gene expression. Expression pattern of the clade D FcSIT (211148) in this double depletion experiment intriguingly suggests its possible specific role for Si uptake during valve formation; this gene expression is significantly induced in only those cells forming the valve (L+S+), as compared those in G1 arrested cells (L+S−). Its responsiveness to Si also disappears under the dark conditions, which arrest the cells at G1. Therefore, the transition of cell cycle to G2/M phase of cells might be somehow correlated to the higher level induction of this gene expression.
CONCLUSION
In this study, we found that the Fc cell cycle can be effectively synchronized by either Si or light. It required approximately 10 h for G1-phase cells to progress to the S-phase and 18 h to reach a peak accumulation at the G2/M phase. The FcSIT multigene family is differentially expressed depending on Si− or light-synchronized Fc cells. Selective member (s) of the FcSIT genes respond to silicate and light, with a different set of genes being responsive to each factor. The Si/light double depletion experiments demonstrated that expression of clade D FcSIT genes is possibly correlated to transition to G2/M phase of the cell cycle, when the valve is actively formed.
Such a diverse responsiveness of SIT gene expression to various environmental cues would provide Fc cells with the capacity for fine-tuning of Si transport in fluctuating environments. In addition to transcriptional responsiveness, the abundance of SIT proteins appears to be regulated at multiple levels, since the levels of SIT proteins are not directly correlated with the abundance of the transcripts (Thamatrakoln and Hildebrand 2007). Therefore, actual protein levels of the specific FcSIT and also their subcellular locations need to be determined to understand the more specific role of each FcSIT in the future.